Integrating the Tumor Microenvironment into Cancer Therapy

 ,
,
Abstract
:1. Introduction
2. Is the TME Potentially Reprogrammable?
3. Emerging Systems of TME Reprogramming
3.1. Intrinsic Systems Capable of Inducing TME Reprogramming
3.1.1. Remodeling TME to Enhance Antitumor Immune Activity
3.1.2. The Nervous System (NS), Adrenaline and Glucocorticoids, and Their Role in Metastases
3.1.3. Intestinal Microbiota as TME Regulator
3.1.4. Metabolic Regulation and Mitochondrial Dysfunction of Cancer
3.1.5. Mechanotransduction and Biotensegrity as Properties of the TME
3.2. Extrinsic Stimuli with the Capacity to Induce TME Reprogramming
3.2.1. Effect of Surgery
3.2.2. Stress, Psychosocial Factors, and Physical Exercise
3.2.3. Vitamin D3 and Vitamin D Receptor (VDR)
3.2.4. Metformin and Other Biguanides
3.2.5. Phytochemicals Derived from Natural Sources
3.2.6. Tensional Homeostasis
4. Evaluation of New Biomarkers
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Riss, J.; Khanna, C.; Koo, S.; Chandramouli, G.V.; Yang, H.H.; Hu, Y.; Kleiner, D.E.; Rosenwald, A.; Schaefer, C.F.; Ben-Sasson, S.A.; et al. Cancers as wounds that do not heal: Differences and similarities between renal regeneration/repair and renal cell carcinoma. Cancer Res. 2006, 66, 7216–7224. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, A. Mechanisms of resistance to EGFR Tyrosine kinase inhibitors and therapeutic approaches: An update. Adv. Exp. Med. Biol. 2016, 893, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. Bioessays 2011, 33, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.T.; Prechtl, A.M.; Pearson, G.W. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res. 2011, 71, 6857–6866. [Google Scholar] [CrossRef] [Green Version]
- Maffini, M.V.; Soto, A.M.; Calabro, J.M.; Ucci, A.A.; Sonnenschein, C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004, 117, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Maffini, M.V.; Calabro, J.M.; Soto, A.M.; Sonnenschein, C. Stromal regulation of neoplastic development: Age-dependent normalization of neoplastic mammary cells by mammary stroma. Am. J. Pathol. 2005, 167, 1405–1410. [Google Scholar] [CrossRef]
- Krause, S.; Maffini, M.V.; Soto, A.M.; Sonnenschein, C. The microenvironment determines the breast cancer cells’ phenotype: Organization of MCF7 cells in 3D cultures. BMC Cancer 2010, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kroemer, G. Cancer cells thrive on stress. Trends Cell Biol. 2019, 29, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.M.; Munst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Weidenfeld, K.; Barkan, D. EMT and stemness in tumor dormancy and outgrowth: Are they intertwined processes? Front. Oncol. 2018, 8, 381. [Google Scholar] [CrossRef] [PubMed]
- van Zijl, F.; Zulehner, G.; Petz, M.; Schneller, D.; Kornauth, C.; Hau, M.; Machat, G.; Grubinger, M.; Huber, H.; Mikulits, W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009, 5, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Roy Choudhury, A.; Gupta, S.; Chaturvedi, P.K.; Kumar, N.; Pandey, D. Mechanobiology of cancer stem cells and their niche. Cancer Microenviron. 2019, 12, 17–27. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef] [Green Version]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Van der Jeught, K.; Bialkowski, L.; Daszkiewicz, L.; Broos, K.; Goyvaerts, C.; Renmans, D.; Van Lint, S.; Heirman, C.; Thielemans, K.; Breckpot, K. Targeting the tumor microenvironment to enhance antitumor immune responses. Oncotarget 2015, 6, 1359–1381. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Drak Alsibai, K.; Meseure, D. Significance of Tumor Microenvironment Scoring and Immune Biomarkers in Patient Stratification and Cancer Outcomes; ItechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Castermans, K.; Griffioen, A.W. Tumor blood vessels, a difficult hurdle for infiltrating leukocytes. Biochim. Biophys. Acta 2007, 1776, 160–174. [Google Scholar] [CrossRef]
- O’Connor, R.S.; Hao, X.; Shen, K.; Bashour, K.; Akimova, T.; Hancock, W.W.; Kam, L.C.; Milone, M.C. Substrate rigidity regulates human T cell activation and proliferation. J. Immunol. 2012, 189, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Zhang, S.; Fan, Y.; Liu, K.; Du, F.; Davey, A.M.; Zhang, H.; Han, W.; Xiong, C.; Liu, W. B cell activation is regulated by the stiffness properties of the substrate presenting the antigens. J. Immunol. 2013, 190, 4661–4675. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Yi, J.; Wan, Z.; Liu, K.; Song, P.; Chau, A.; Wang, F.; Chang, Z.; Han, W.; Zheng, W.; et al. Substrate stiffness regulates B-cell activation, proliferation, class switch, and T-cell-independent antibody responses in vivo. Eur. J. Immunol. 2015, 45, 1621–1634. [Google Scholar] [CrossRef]
- Huse, M. Mechanical forces in the immune system. Nat. Rev. Immunol. 2017, 17, 679–690. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The dark side of fibroblasts: Cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Itano, N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016, 375, 20–30. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Falk, B.A.; Wu, R.P.; Buckner, J.H.; Wight, T.N.; Nepom, G.T. Intact extracellular matrix and the maintenance of immune tolerance: High molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 2009, 86, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, T.; Bromberg, J.S. Regulation of the Immune System by Laminins. Trends Immunol. 2017, 38, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.H.; Edelman, E.R.; Stultz, C.M. Collagen fragments modulate innate immunity. Exp. Biol. Med. (Maywood) 2007, 232, 406–411. [Google Scholar]
- Horejs, C.M.; Serio, A.; Purvis, A.; Gormley, A.J.; Bertazzo, S.; Poliniewicz, A.; Wang, A.J.; DiMaggio, P.; Hohenester, E.; Stevens, M.M. Biologically-active laminin-111 fragment that modulates the epithelial-to-mesenchymal transition in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 5908–5913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L. Classification of Advanced Human Cancers Based on Tumor Immunity in the MicroEnvironment (TIME) for Cancer Immunotherapy. JAMA Oncol. 2016, 2, 1403–1404. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014, 20, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Lim, S.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis Oncol. 2019, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondicova, K.; Mravec, B. Role of nervous system in cancer aetiopathogenesis. Lancet Oncol. 2010, 11, 596–601. [Google Scholar] [CrossRef]
- Noguera, R.; Burgos-Panadero, R.; Gamero-Sandemetrio, E.; de la Cruz-Merino, L.; Alvaro Naranjo, T. An integral view of cancer (II). Fields of investigation and emerging biomarkers. Rev. Esp. Patol. 2019, 52, 222–233. [Google Scholar] [CrossRef]
- Green McDonald, P.; O’Connell, M.; Lutgendorf, S.K. Psychoneuroimmunology and cancer: A decade of discovery, paradigm shifts, and methodological innovations. Brain Behav. Immun. 2013, 30, S1–S9. [Google Scholar] [CrossRef] [Green Version]
- Saloman, J.L.; Albers, K.M.; Rhim, A.D.; Davis, B.M. Can Stopping Nerves, Stop Cancer? Trends Neurosci. 2016, 39, 880–889. [Google Scholar] [CrossRef] [Green Version]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascal, D. Tumor progression: The neuronal input. Ann. Transl. Med. 2018, 6, 89. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Entschladen, F.; Drell, T.L.t.; Lang, K.; Joseph, J.; Zaenker, K.S. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004, 5, 254–258. [Google Scholar] [CrossRef]
- Mancino, M.; Ametller, E.; Gascon, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta 2011, 1816, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.M.; Hayakawa, Y.; Kodama, Y.; Muthupalani, S.; Westphalen, C.B.; Andersen, G.T.; Flatberg, A.; Johannessen, H.; Friedman, R.A.; Renz, B.W.; et al. Denervation suppresses gastric tumorigenesis. Sci. Transl. Med. 2014, 6, 250ra115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deborde, S.; Omelchenko, T.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.F.; Chernichenko, N.; Lee, S.Y.; Barajas, F.; Chen, C.H.; et al. Schwann cells induce cancer cell dispersion and invasion. J. Clin. Investig. 2016, 126, 1538–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.Y.; De Paz, D.; Lin, C.Y.; Fan, K.H.; Wang, H.M.; Hsieh, C.H.; Lee, L.A.; Yen, T.C.; Liao, C.T.; Yeh, C.H.; et al. Prognostic impact of extratumoral perineural invasion in patients with oral cavity squamous cell carcinoma. Cancer Med. 2019, 8, 6185–6194. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Shi, S.; Xu, J.; Zhang, B.; Qin, Y.; Ji, S.; Xu, W.; Liu, J.; Liu, L.; Liu, C.; et al. New insights into perineural invasion of pancreatic cancer: More than pain. Biochim. Biophys. Acta 2016, 1865, 111–122. [Google Scholar] [CrossRef]
- Deng, J.; You, Q.; Gao, Y.; Yu, Q.; Zhao, P.; Zheng, Y.; Fang, W.; Xu, N.; Teng, L. Prognostic value of perineural invasion in gastric cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e88907. [Google Scholar] [CrossRef] [Green Version]
- Mirkin, K.A.; Hollenbeak, C.S.; Mohamed, A.; Jia, Y.; El-Deiry, W.S.; Messaris, E. Impact of perineural invasion on survival in node negative colon cancer. Cancer Biol. Ther. 2017, 18, 740–745. [Google Scholar] [CrossRef]
- Eng, J.W.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: The impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol. Immunother. 2014, 63, 1115–1128. [Google Scholar] [CrossRef] [Green Version]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the nervous system in cancer metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 5. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, P.J. Is norepinephrine an etiological factor in some types of cancer? Int. J. Cancer 2009, 124, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Sun, S.P.; Wu, J.I.; Wang, L.H. Low-dose glucocorticoids suppresses ovarian tumor growth and metastasis in an immunocompetent syngeneic mouse model. PLoS ONE 2017, 12, e0178937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer 2015, 15, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Cai, X.; Zhang, J.; Wang, W.; Sheng, Q.; Hua, H.; Zhou, X. Role of Gut Microbiota in the Development and Treatment of Colorectal Cancer. Digestion 2019, 100, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271–285. [Google Scholar] [CrossRef]
- Adachi, K.; Tamada, K. Microbial biomarkers for immune checkpoint blockade therapy against cancer. J. Gastroenterol. 2018, 53, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Meng, S.; Chen, B.; Yang, J.; Wang, J.; Zhu, D.; Meng, Q.; Zhang, L. Study of microbiomes in aseptically collected samples of human breast tissue using needle biopsy and the potential role of in situ tissue microbiomes for promoting malignancy. Front. Oncol. 2018, 8, 318. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Chen, J.; Yao, H.; Hu, H. Fusobacterium and colorectal cancer. Front. Oncol. 2018, 8, 371. [Google Scholar] [CrossRef]
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The brain-gut-microbiome axis. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Sun, C.H.; Li, B.B.; Wang, B.; Zhao, J.; Zhang, X.Y.; Li, T.T.; Li, W.B.; Tang, D.; Qiu, M.J.; Wang, X.C.; et al. The role of Fusobacterium nucleatum in colorectal cancer: From carcinogenesis to clinical management. Chronic Dis. Transl. Med. 2019, 5, 178–187. [Google Scholar] [CrossRef]
- Zitvogel, L.; Daillere, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef]
- Yi, M.; Yu, S.; Qin, S.; Liu, Q.; Xu, H.; Zhao, W.; Chu, Q.; Wu, K. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Yang, L.; Xu, Q.; Yuan, H.; Wang, W.; Xia, W.; Gong, D.; Zhang, W.; Yu, K. CD68- and CD163-positive tumor infiltrating macrophages in non-metastatic breast cancer: A retrospective study and meta-analysis. J. Cancer 2019, 10, 4463–4472. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, S.; Tanaka, H.; Nishimura, J.; Sakimura, C.; Tamura, T.; Toyokawa, T.; Muguruma, K.; Yashiro, M.; Hirakawa, K.; Ohira, M. Neutrophils in primary gastric tumors are correlated with neutrophil infiltration in tumor-draining lymph nodes and the systemic inflammatory response. BMC Immunol. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Weyer-Elberich, V.; Hengstler, J.G.; Heimes, A.S.; Almstedt, K.; Gerhold-Ay, A.; Lebrecht, A.; Battista, M.J.; Hasenburg, A.; Sahin, U.; et al. Prognostic impact of CD4-positive T cell subsets in early breast cancer: A study based on the FinHer trial patient population. Breast Cancer Res. 2018, 20, 15. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannani, D.; Vétizou, M.; Enot, D.; Rusakiewicz, S.; Chaput, N.; Klatzmann, D.; Desbois, M.; Jacquelot, N.; Vimond, N.; Chouaib, S.; et al. Anticancer immunotherapy by CTLA-4 blockade: Obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef]
- deLeeuw, R.J.; Kost, S.E.; Kakal, J.A.; Nelson, B.H. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: A critical review of the literature. Clin. Cancer Res. 2012, 18, 3022–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiffoleau, E. C-Type Lectin-Like Receptors As emerging orchestrators of sterile inflammation represent potential therapeutic targets. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef]
- Edin, S.; Kaprio, T.; Hagström, J.; Larsson, P.; Mustonen, H.; Böckelman, C.; Strigård, K.; Gunnarsson, U.; Haglund, C.; Palmqvist, R. The prognostic importance of CD20+ B lymphocytes in Colorectal cancer and the relation to other immune cell subsets. Sci. Rep. 2019, 9, 19997. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Park, C.K.; Jung, W.H.; Koo, J.S. Expression of cancer-associated fibroblast-related proteins differs between invasive lobular carcinoma and invasive ductal carcinoma. Breast Cancer Res. Treat. 2016, 159, 55–69. [Google Scholar] [CrossRef]
- Iwamoto, S.; Burrows, R.C.; Agoff, S.N.; Piepkorn, M.; Bothwell, M.; Schmidt, R. The p75 neurotrophin receptor, relative to other Schwann cell and melanoma markers, is abundantly expressed in spindled melanomas. Am. J. Dermatopathol. 2001, 23, 288–294. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Zhang, X. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadeo, I.; Berbegall, A.P.; Navarro, S.; Castel, V.; Noguera, R. A stiff extracellular matrix is associated with malignancy in peripheral neuroblastic tumors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Berdiaki, A.; Spyridaki, I.; Krasanakis, T.; Tsatsakis, A.; Tzanakakis, G.N. Proteoglycans-biomarkers and targets in cancer therapy. Front. Endocrinol. (Lausanne) 2018, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Kondisetty, S.; Menon, K.N.; Pooleri, G.K. Fibronectin protein expression in renal cell carcinoma in correlation with clinical stage of tumour. Biomark. Res. 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Rousselle, P.; Scoazec, J.Y. Laminin 332 in cancer: When the extracellular matrix turns signals from cell anchorage to cell movement. Semin. Cancer Biol. 2020, 62, 149–165. [Google Scholar] [CrossRef]
- Burgos-Panadero, R.; Noguera, I.; Cañete, A.; Navarro, S.; Noguera, R. Vitronectin as a molecular player of the tumor microenvironment in neuroblastoma. BMC Cancer 2019, 19, 479. [Google Scholar] [CrossRef]
- Xu, Y.; Pasche, B. TGF-β signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, R14–R20. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef]
- Vicente-Munuera, P.; Burgos-Panadero, R.; Noguera, I.; Navarro, S.; Noguera, R.; Escudero, L.M. The topology of vitronectin: A complementary feature for neuroblastoma risk classification based on computer-aided detection. Int. J. Cancer 2020, 146, 553–565. [Google Scholar] [CrossRef]
- Tadeo, I.; Berbegall, A.P.; Escudero, L.M.; Alvaro, T.; Noguera, R. Biotensegrity of the extracellular matrix: Physiology, dynamic mechanical balance, and implications in oncology and mechanotherapy. Front. Oncol. 2014, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular mechanotransduction: From tension to function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Croteau, E.; Renaud, J.M.; Richard, M.A.; Ruddy, T.D.; Bénard, F.; deKemp, R.A. PET Metabolic biomarkers for cancer. Biomark. Cancer 2016, 8, 61–69. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C.; Shen, Z.; Zhang, X.; Chen, Y.; Lin, F.; Ma, X.; Zhuang, C.; Mao, Y.; Gan, H.; et al. Extracellular pH is a biomarker enabling detection of breast cancer and liver cancer using CEST MRI. Oncotarget 2017, 8, 45759–45767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 alpha impacts on their stability and mobility. Open Biol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faes, S.; Uldry, E.; Planche, A.; Santoro, T.; Pythoud, C.; Demartines, N.; Dormond, O. Acidic pH reduces VEGF-mediated endothelial cell responses by downregulation of VEGFR-2; relevance for anti-angiogenic therapies. Oncotarget 2016, 7, 86026–86038. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, J.; Guan, Y.; Lou, Y.; Chen, H.; Xu, M.; Deng, D.; Chen, J.; Ni, B.; Zhao, L.; et al. Dysbiosis of the Gut microbiome is associated with tumor biomarkers in lung cancer. Int. J. Biol. Sci. 2019, 15, 2381–2392. [Google Scholar] [CrossRef]
- Leinwand, J.C.; Miller, G. Microbes as biomarkers and targets in pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 16, 665–666. [Google Scholar] [CrossRef]
- Shirazi, M.S.R.; Al-Alo, K.Z.K.; Al-Yasiri, M.H.; Lateef, Z.M.; Ghasemian, A. Microbiome dysbiosis and predominant bacterial species as human cancer biomarkers. J. Gastrointest. Cancer 2019. [Google Scholar] [CrossRef]
- Hoeppner, L.H.; Wang, Y.; Sharma, A.; Javeed, N.; Van Keulen, V.P.; Wang, E.; Yang, P.; Roden, A.C.; Peikert, T.; Molina, J.R.; et al. Dopamine D2 receptor agonists inhibit lung cancer progression by reducing angiogenesis and tumor infiltrating myeloid derived suppressor cells. Mol. Oncol. 2015, 9, 270–281. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ru, G.Q.; Ma, Y.Y.; Xie, J.; Chen, W.Y.; Wang, H.J.; Wang, S.B.; Li, L.; Jin, K.T.; He, X.L.; et al. High expression of substance P and its receptor neurokinin-1 receptor in colorectal cancer is associated with tumor progression and prognosis. Onco. Targets Ther. 2016, 9, 3595–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Murugan, S.; Boyadjieva, N.; Jabbar, S.; Shrivastava, P.; Sarkar, D.K. Beta-endorphin cell therapy for cancer prevention. Cancer Prev. Res. (Phila.) 2015, 8, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.; Hebrant, A.; Dumont, J.E. Metabolic reprogramming of the tumor. Oncogene 2012, 31, 3999–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic switch in the tumor microenvironment determines immune responses to anti-cancer therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Schwartsburd, P. Cancer-induced reprogramming of host glucose metabolism: “vicious cycle” supporting cancer progression. Front. Oncol. 2019, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Ko, Y.H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Biol. Ther. 2011, 12, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivadeneira, D.B.; Delgoffe, G.M. Antitumor T-cell reconditioning: Improving metabolic fitness for optimal cancer immunotherapy. Clin. Cancer Res. 2018, 24, 2473–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 403. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P.; Creighton, C.J.; Chan, W.Y.; Wong, L.C. Correction: Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS ONE 2019, 14, e0221671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Sharma, L.K.; Bai, Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009, 19, 802–815. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer: Warburg addressed. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Alvaro, T.; de la Cruz-Merino, L.; Henao-Carrasco, F.; Villar Rodriguez, J.L.; Vicente Baz, D.; Codes Manuel de Villena, M.; Provencio, M. Tumor microenvironment and immune effects of antineoplastic therapy in lymphoproliferative syndromes. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical forces in tumor angiogenesis. Adv. Exp. Med. Biol. 2018, 1092, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.M.; Burridge, K. Mechanotransduction and nuclear function. Curr. Opin. Cell Biol. 2016, 40, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Rianna, C.; Kumar, P.; Radmacher, M. The role of the microenvironment in the biophysics of cancer. Semin. Cell Dev. Biol. 2018, 73, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Panadero, R.; Lucantoni, F.; Gamero-Sandemetrio, E.; Cruz-Merino, L.; Alvaro, T.; Noguera, R. The tumour microenvironment as an integrated framework to understand cancer biology. Cancer Lett. 2019, 461, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.; Hanna, M.G.; Geneslaw, L.; Miraflor, A.; Werneck Krauss Silva, V.; Busam, K.J.; Brogi, E.; Reuter, V.E.; Klimstra, D.S.; Fuchs, T.J. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 2019, 25, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Johnson, J.; Mollaee, M.; Tassone, P.; Amin, D.; Knops, A.; Whitaker-Menezes, D.; Mahoney, M.G.; South, A.; Rodeck, U.; et al. Metformin clinical trial in HPV+ and HPV- head and neck squamous cell carcinoma: Impact on cancer cell apoptosis and immune infiltrate. Front. Oncol. 2018, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.; Butler, D.; El Haj, A.; Bodle, J.C.; Loboa, E.G.; Banes, A.J. Key developments that impacted the field of mechanobiology and mechanotransduction. J. Orthop. Res. 2018, 36, 605–619. [Google Scholar] [CrossRef]
- Li, L.; Eyckmans, J.; Chen, C.S. Designer biomaterials for mechanobiology. Nat. Mater. 2017, 16, 1164–1168. [Google Scholar] [CrossRef]
- Neeman, E.; Ben-Eliyahu, S. Surgery and stress promote cancer metastasis: New outlooks on perioperative mediating mechanisms and immune involvement. Brain Behav. Immun. 2013, 30, S32–S40. [Google Scholar] [CrossRef] [Green Version]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, J.; Yamada, T.; Egashira, N.; Ueda, M.; Zukeyama, N.; Ushio, S.; Masuda, S. Comparison of the anti-tumor effects of selective serotonin reuptake inhibitors as well as serotonin and norepinephrine reuptake inhibitors in human hepatocellular carcinoma Cells. Biol. Pharm. Bull. 2015, 38, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannen, V.; Garcia, S.B.; Silva, W.A., Jr.; Gasser, M.; Monch, R.; Alho, E.J.; Heinsen, H.; Scholz, C.J.; Friedrich, M.; Heinze, K.G.; et al. Oncostatic effects of fluoxetine in experimental colon cancer models. Cell Signal. 2015, 27, 1781–1788. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Casado, A.; Martin-Ruiz, A.; Perez, L.M.; Provencio, M.; Fiuza-Luces, C.; Lucia, A. Exercise and the hallmarks of cancer. Trends Cancer 2017, 3, 423–441. [Google Scholar] [CrossRef]
- Idorn, M.; Thor Straten, P. Exercise and cancer: From “healthy” to “therapeutic”? Cancer Immunol. Immunother. 2017, 66, 667–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormie, P.; Zopf, E.M.; Zhang, X.; Schmitz, K.H. The Impact of exercise on cancer mortality, recurrence, and treatment-related adverse effects. Epidemiol. Rev. 2017, 39, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Kim, J.J.; Mohr, S.B.; Gorham, E.D.; Grant, W.B.; Giovannucci, E.L.; Baggerly, L.; Hofflich, H.; Ramsdell, J.W.; Zeng, K.; et al. Meta-analysis of all-cause mortality according to serum 25-hydroxyvitamin D. Am. J. Public Health 2014, 104, e43–e50. [Google Scholar] [CrossRef] [PubMed]
- Maalmi, H.; Walter, V.; Jansen, L.; Boakye, D.; Schottker, B.; Hoffmeister, M.; Brenner, H. Association between blood 25-Hydroxyvitamin D levels and survival in colorectal cancer patients: An updated systematic review and meta-analysis. Nutrients 2018, 10, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Baggerly, L.L.; Garland, C.F.; Gorham, E.D.; Hollis, B.W.; Trump, D.L.; Lappe, J.M. Breast cancer risk markedly lower with serum 25-hydroxyvitamin D concentrations >/=60 vs. <20 ng/ml (150 vs. 50 nmol/L): Pooled analysis of two randomized trials and a prospective cohort. PLoS ONE 2018, 13, e0199265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haznadar, M.; Krausz, K.W.; Margono, E.; Diehl, C.M.; Bowman, E.D.; Manna, S.K.; Robles, A.I.; Ryan, B.M.; Gonzalez, F.J.; Harris, C.C. Inverse association of vitamin D3 levels with lung cancer mediated by genetic variation. Cancer Med. 2018, 7, 2764–2775. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.L.; Baggerly, C.; French, C.B.; Baggerly, L.L.; Garland, C.F.; Gorham, E.D.; Lappe, J.M.; Heaney, R.P. Serum 25-Hydroxyvitamin D concentrations >/=40 ng/ml are associated with >65% lower cancer Risk: Pooled analysis of randomized trial and prospective cohort Study. PLoS ONE 2016, 11, e0152441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, P.J.; Marcinkowska, E.; Gocek, E.; Studzinski, G.P.; Brown, G. Vitamin D3-driven signals for myeloid cell differentiation--implications for differentiation therapy. Leuk. Res. 2010, 34, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, J.H.; Li, Y.; Rogers, C.J.; Cantorna, M.T. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J. Nutr. 2013, 143, 1679–1686. [Google Scholar] [CrossRef]
- Lee, T.Y.; Martinez-Outschoorn, U.E.; Schilder, R.J.; Kim, C.H.; Richard, S.D.; Rosenblum, N.G.; Johnson, J.M. Metformin as a therapeutic target in endometrial cancers. Front. Oncol. 2018, 8, 341. [Google Scholar] [CrossRef]
- Ackerman, S.E.; Blackburn, O.A.; Marchildon, F.; Cohen, P. Insights into the link between obesity and cancer. Curr. Obes. Rep. 2017, 6, 195–203. [Google Scholar] [CrossRef]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4(+)CD25(+) regulatory T Cells in the tumor microenvironment by metformin, a Type 2 Diabetes drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef]
- Tang, D.; Zhang, S.; Shi, X.; Wu, J.; Yin, G.; Tan, X.; Liu, F.; Wu, X.; Du, X. Combination of Astragali Polysaccharide and curcumin improves the morphological structure of tumor vessels and induces tumor vascular normalization to inhibit the growth of hepatocellular carcinoma. Integr. Cancer Ther. 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Kay, N.E.; Secreto, C.R.; Shanafelt, T.D. Curcumin inhibits prosurvival pathways in chronic lymphocytic leukemia B cells and may overcome their stromal protection in combination with EGCG. Clin. Cancer Res. 2009, 15, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudas, J.; Fullar, A.; Romani, A.; Pritz, C.; Kovalszky, I.; Hans Schartinger, V.; Mathias Sprinzl, G.; Riechelmann, H. Curcumin targets fibroblast-tumor cell interactions in oral squamous cell carcinoma. Exp. Cell Res. 2013, 319, 800–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeini, M.B.; Momtazi, A.A.; Jaafari, M.R.; Johnston, T.P.; Barreto, G.; Banach, M.; Sahebkar, A. Antitumor effects of curcumin: A lipid perspective. J. Cell Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ichikawa, H.; Takada, Y.; Sandur, S.K.; Shishodia, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates expression of cell proliferation and antiapoptotic and metastatic gene products through suppression of IkappaBalpha kinase and Akt activation. Mol. Pharmacol. 2006, 69, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Hsu, C.H.; Lin, J.K.; Hsu, M.M.; Ho, Y.F.; Shen, T.S.; Ko, J.Y.; Lin, J.T.; Lin, B.R.; Ming-Shiang, W.; et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001, 21, 2895–2900. [Google Scholar]
- Wong, S.W.; Lenzini, S.; Shin, J.W. Perspective: Biophysical regulation of cancerous and normal blood cell lineages in hematopoietic malignancies. APL Bioeng. 2018, 2, 031802. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.L.; Yalavarthy, P.K.; Doyley, M.M.; Dehghani, H.; Pogue, B.W. Interstitial fluid pressure in soft tissue as a result of an externally applied contact pressure. Phys. Med. Biol. 2007, 52, 4121–4136. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Barbone, P.E.; Smith, M.L.; Stamenovic, D. Effect of correlation between traction forces on tensional homeostasis in clusters of endothelial cells and fibroblasts. J. Biomech. 2020, 100, 109588. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Dunn, A.R.; Moses, M.A.; Van Vliet, K.J. Endothelial cells as mechanical transducers: Enzymatic activity and network formation under cyclic strain. Mech. Chem. Biosyst. 2004, 1, 279–290. [Google Scholar] [PubMed]
- Bisson, M.A.; Beckett, K.S.; McGrouther, D.A.; Grobbelaar, A.O.; Mudera, V. Transforming growth factor-beta1 stimulation enhances Dupuytren’s fibroblast contraction in response to uniaxial mechanical load within a 3-dimensional collagen gel. J. Hand Surg. Am. 2009, 34, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Mao, Z.; Peng, Z.; Zhou, L.; Chen, Y.; Huang, P.H.; Truica, C.I.; Drabick, J.J.; El-Deiry, W.S.; Dao, M.; et al. Acoustic separation of circulating tumor cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4970–4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Huang, P.H.; Zhang, R.; Mao, Z.; Chen, C.; Kemeny, G.; Li, P.; Lee, A.V.; Gyanchandani, R.; Armstrong, A.J.; et al. Circulating Tumor Cell Phenotyping via High-Throughput Acoustic Separation. Small 2018, 14, e1801131. [Google Scholar] [CrossRef]
- Islam, M.; Mezencev, R.; McFarland, B.; Brink, H.; Campbell, B.; Tasadduq, B.; Waller, E.K.; Lam, W.; Alexeev, A.; Sulchek, T. Microfluidic cell sorting by stiffness to examine heterogenic responses of cancer cells to chemotherapy. Cell Death Dis. 2018, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhao, F.; Li, Z.; Yu, J. Current landscape and future directions of biomarkers for predicting responses to immune checkpoint inhibitors. Cancer Manag. Res. 2018, 10, 2475–2488. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. Cancer immunology. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; Lopez-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Senovilla, L.; Vacchelli, E.; Galon, J.; Adjemian, S.; Eggermont, A.; Fridman, W.H.; Sautes-Fridman, C.; Ma, Y.; Tartour, E.; Zitvogel, L.; et al. Trial watch: Prognostic and predictive value of the immune infiltrate in cancer. Oncoimmunology 2012, 1, 1323–1343. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Immune responses to cancer: Are they potential biomarkers of prognosis? Front. Oncol. 2013, 3, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 2020, 9, 1682381. [Google Scholar] [CrossRef] [Green Version]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. Author correction: High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 1773–1775. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subrahmanyam, P.B.; Dong, Z.; Gusenleitner, D.; Giobbie-Hurder, A.; Severgnini, M.; Zhou, J.; Manos, M.; Eastman, L.M.; Maecker, H.T.; Hodi, F.S. Distinct predictive biomarker candidates for response to anti-CTLA-4 and anti-PD-1 immunotherapy in melanoma patients. J. Immunother. Cancer 2018, 6, 18. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Behren, A.; Thompson, E.W.; Anderson, R.L.; Ferrao, P.T. Editorial: Cancer plasticity and the microenvironment: Implications for immunity and therapy response. Front. Oncol. 2019, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From big data to precision medicine. Front. Med. (Lausanne) 2019, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- D’Alterio, C.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020, 60, 351–361. [Google Scholar] [CrossRef]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for cancer: A trigger for metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]





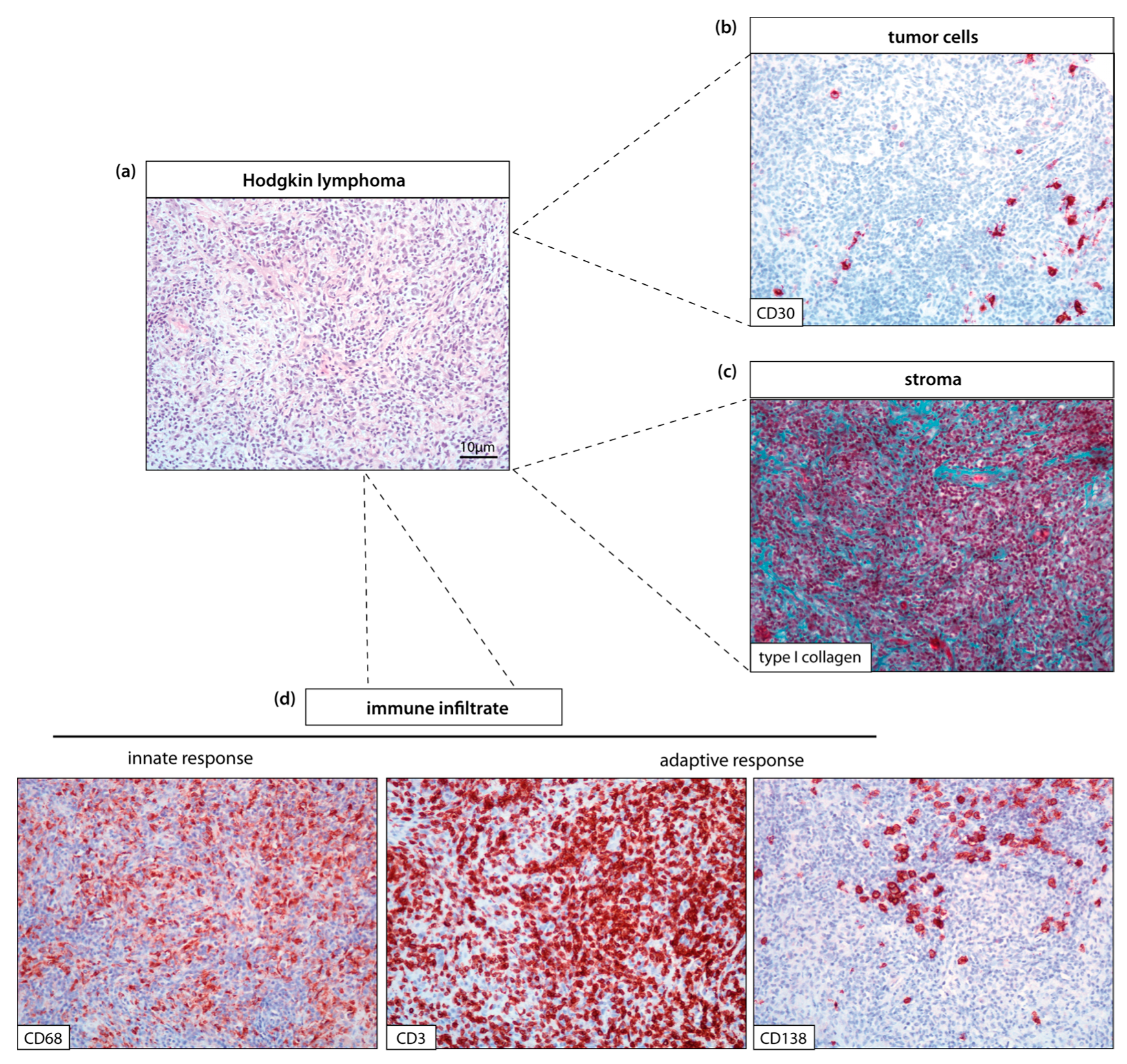{kind=link}
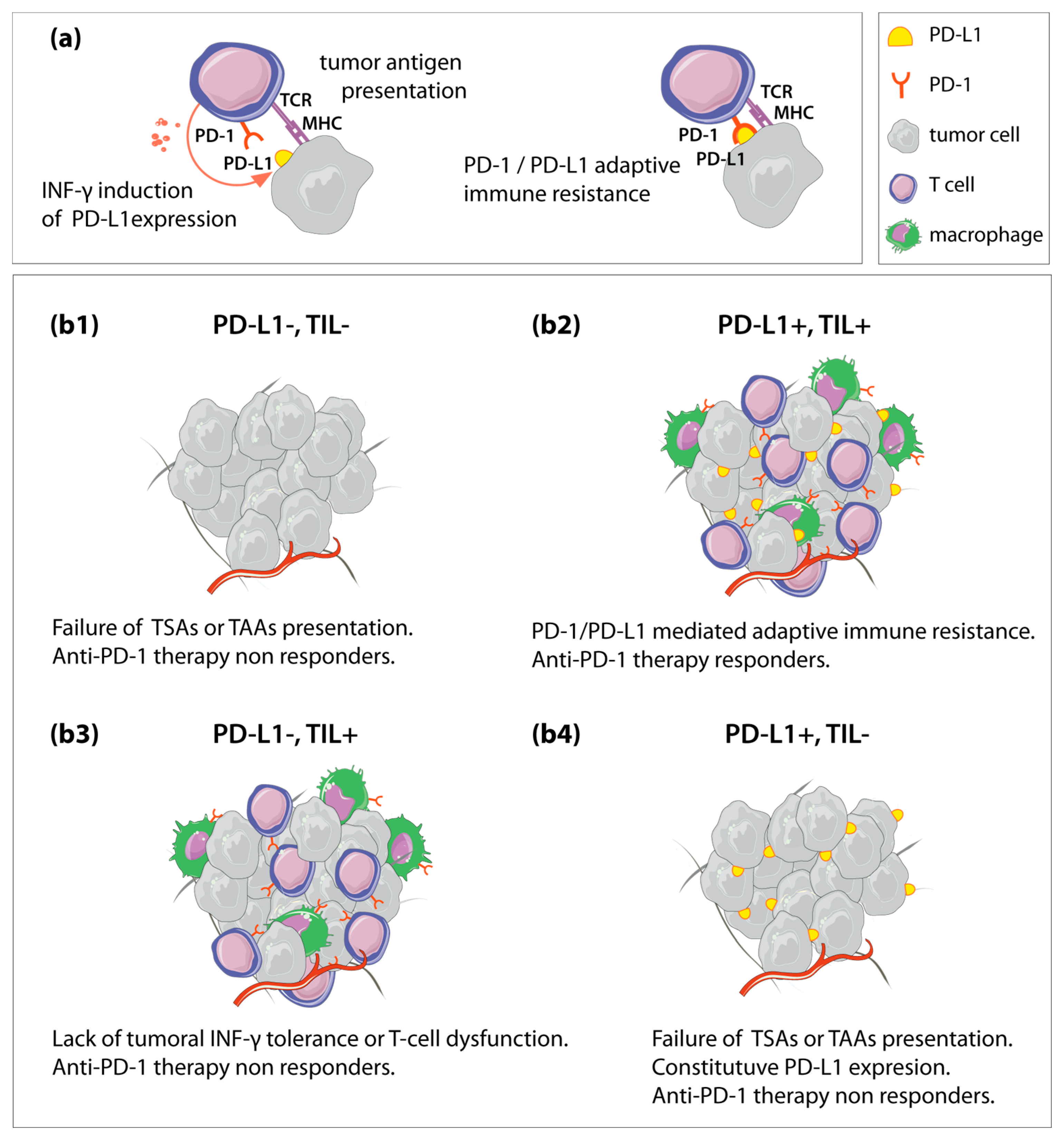{kind=link}
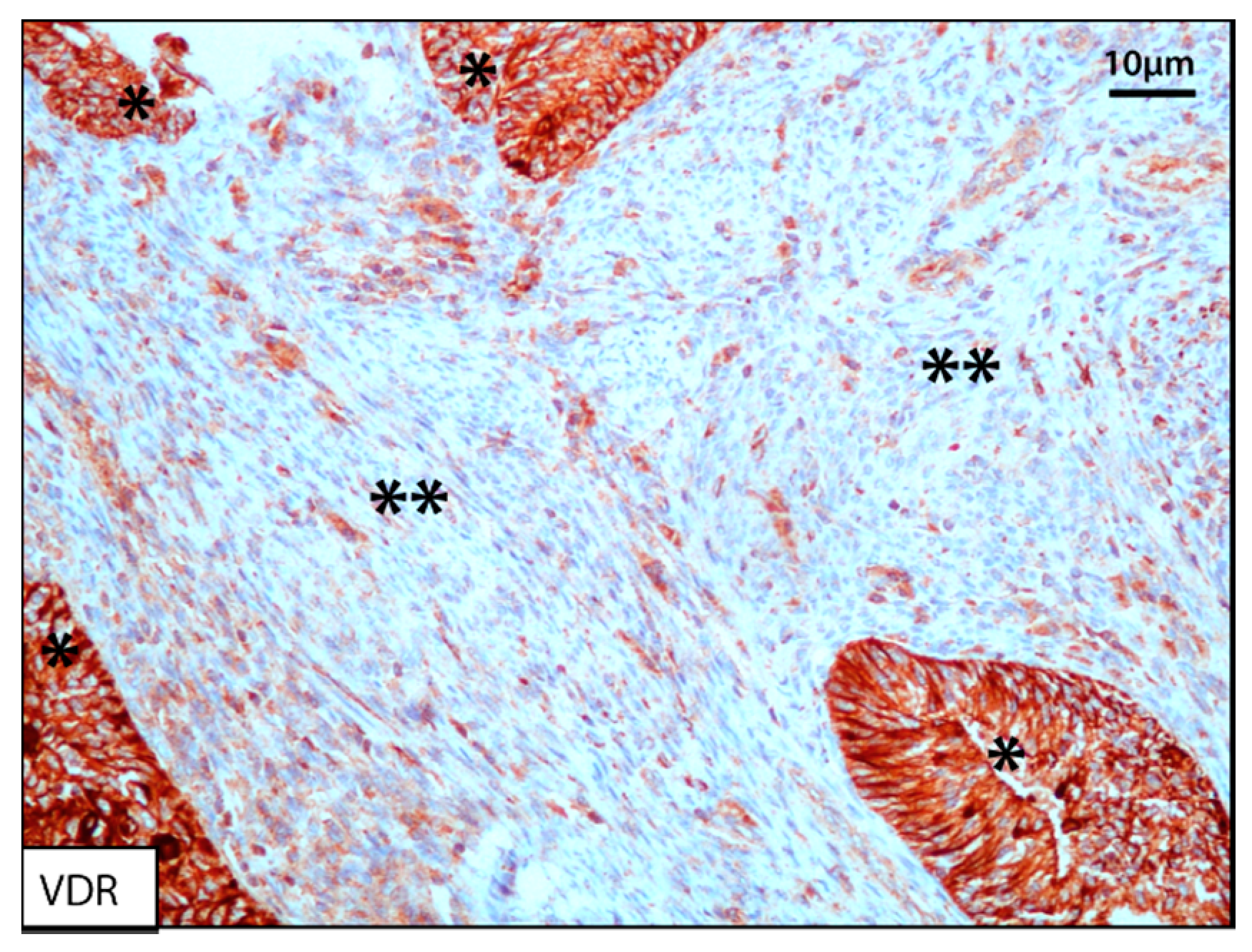{kind=link}
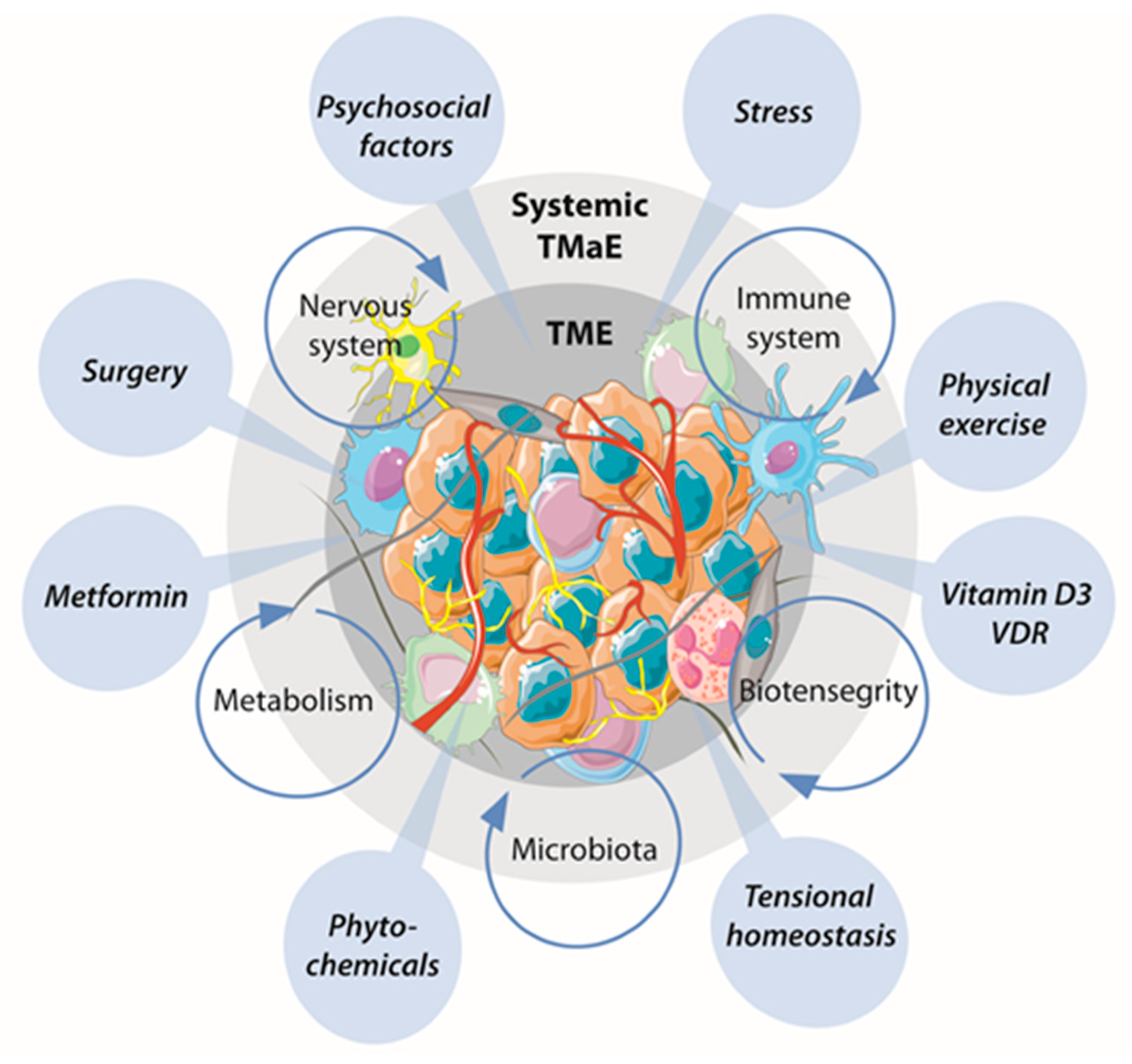{kind=link}
| Evaluation Groups | Parameters | Indicators | Detection | Method | References |
|---|---|---|---|---|---|
| TME | Inflammatory infiltrated cells | TAM | CD68, CD163 | IHC | [79] |
| TAN | CD15, CD32, CD35 | [80] | |||
| T helper | CD4 | [81] | |||
| Cytotoxic T cells | CD8 | [82] | |||
| Memory T cells | CD8, CD4, CTLA-4 | [83] | |||
| Tregs | FOXP3, CD4, CD25 | [84] | |||
| DC | CD141, CLEC9, CD11c | [85,86] | |||
| NK | CD16, CD56, PD-L1, PD-L2 | [87] | |||
| B lymphocytes | CD20 | [88] | |||
| Stromal cells | CAF | αSAM, CD10, FSP1, AEBP1 | [89,90] | ||
| Schwann cells | S100, GFAP, p75NTR | [91] | |||
| MSC | SC | Sox2, Oct4, CD133, Nestin, c-kit | [92] | ||
| Fibers | Collagen type I | T. Masson, van Gieson | HC | [93,94] | |
| Collagen type III | [94] | ||||
| Elastic fibers | Orcein, Gomori, Snook, Wilder, Verhoeff | [94] | |||
| Interstitial fluid | Proteoglycans | Alcian blue | [95] | ||
| Fibronectin | Antifibronectin | IHC | [96] | ||
| Laminin | Antilaminin | [97] | |||
| Vitronectin | Antivitronectin | [98] | |||
| Growth factor | TGF-β | AS | [99] | ||
| Cytokines | Lymphokines | ELISA | [100] | ||
| Proteases | Metalloproteinases | [100] | |||
| Oxygen (ROS) | GSH/GSSG | [100] | |||
| 3D structure | Fibres and Cellular elements | Topology | Graph theory | [101,102] | |
| Mechanical forces | Focal adhesions | (F-actin, myosin II, α-actin, fascin) | IF | [103] | |
| Stress fibres | [103] | ||||
| Mechanotransduction | Mechano-actuated shuttling proteins | (β-catenin, zyxin) | [103] | ||
| LINC complex | (SUN and nesprins) | [103] | |||
| Systemic factors | Glycolic index | ↑metabolic index | 18FDG | PET | [104] |
| pH | Acidosis | Electrolytes serum concentration | Enzymatic | [105] | |
| Oxygen saturation | Hypoxia | ↑HIF-1, ↑lactate | IHC | [106] | |
| Metabolism | Inflammatory response | ↓VEGF | AS, ELISA | [107] | |
| Intestinal microbiota | Dysbiosis | ↑Bacteroids | MALDI-TOF MS | [76,108,109,110] | |
| ↑Fusobacterium | NGS | [76,108,109,110] | |||
| ↑Porphyromonas | 16S rRNA | [76,108,109,110] | |||
| ↑Enterobacter | [76,108,109,110] | ||||
| ↑Cybrobacter | [76,108,109,110] | ||||
| Nervous system | Deregulation | ↑Norepinephrine, | HPLC | [62] | |
| ↑Dopamine | [111] | ||||
| ↑substance P | [112] | ||||
| ↓β-endorphins | [113] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanegre, S.; Lucantoni, F.; Burgos-Panadero, R.; de La Cruz-Merino, L.; Noguera, R.; Álvaro Naranjo, T. Integrating the Tumor Microenvironment into Cancer Therapy. Cancers 2020, 12, 1677. https://doi.org/10.3390/cancers12061677
Sanegre S, Lucantoni F, Burgos-Panadero R, de La Cruz-Merino L, Noguera R, Álvaro Naranjo T. Integrating the Tumor Microenvironment into Cancer Therapy. Cancers. 2020; 12(6):1677. https://doi.org/10.3390/cancers12061677
Chicago/Turabian StyleSanegre, Sabina, Federico Lucantoni, Rebeca Burgos-Panadero, Luis de La Cruz-Merino, Rosa Noguera, and Tomás Álvaro Naranjo. 2020. "Integrating the Tumor Microenvironment into Cancer Therapy" Cancers 12, no. 6: 1677. https://doi.org/10.3390/cancers12061677