The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
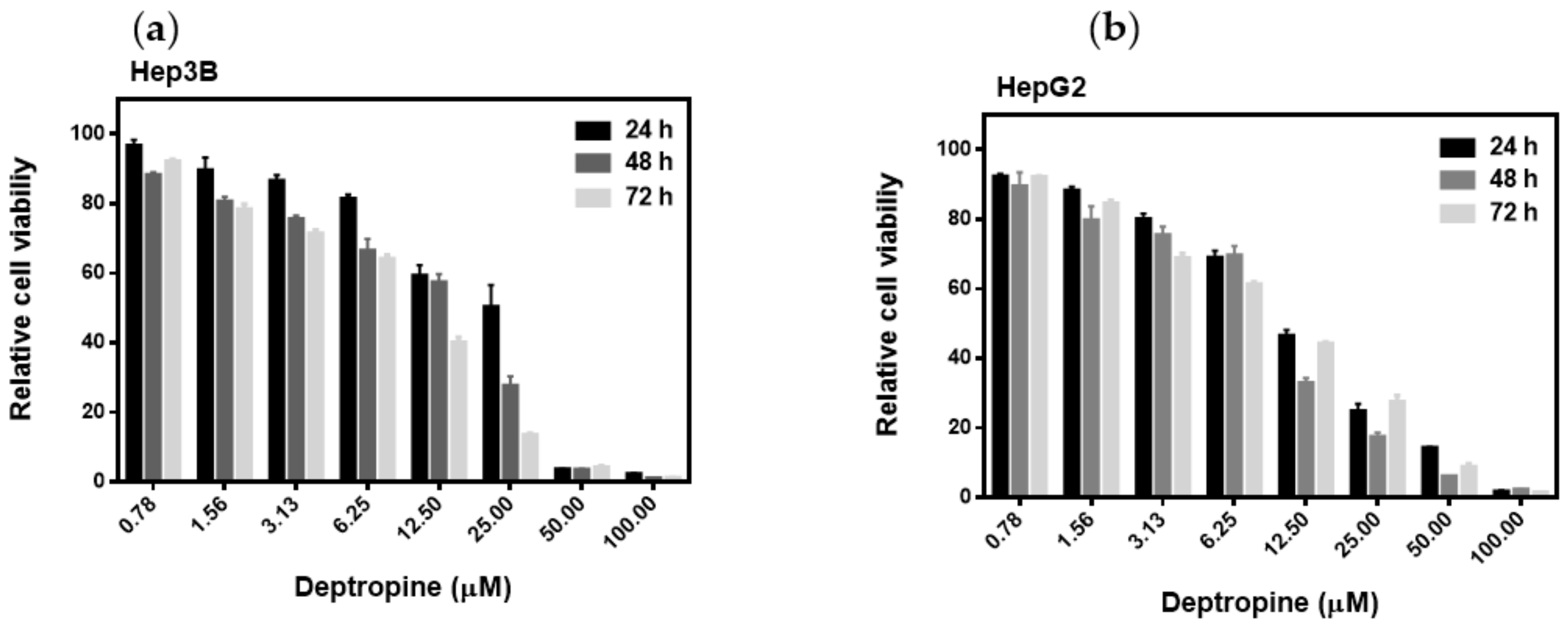
2.1. Deptropine Was the Most Effective Drug against Hepatoma Cell Proliferation
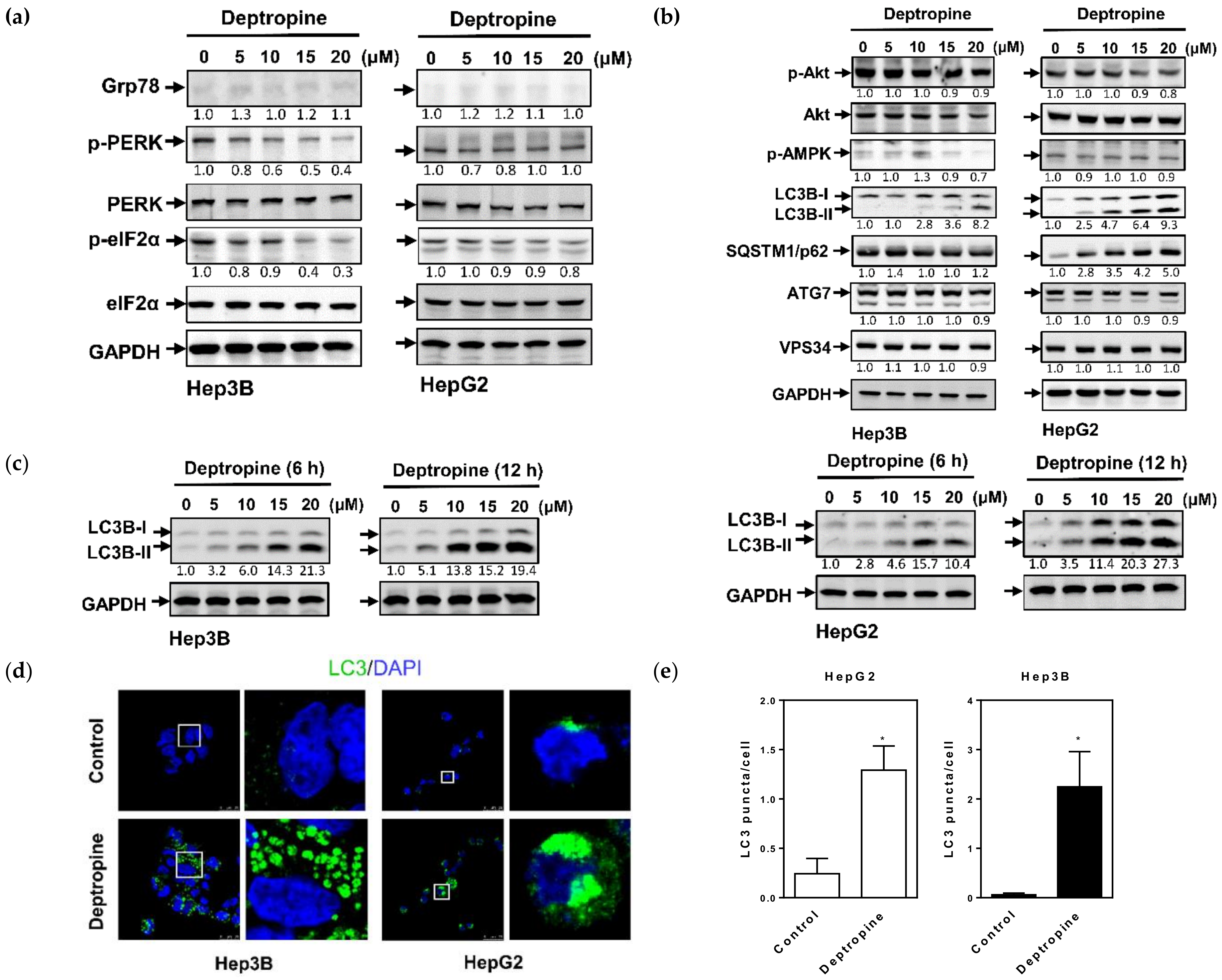
2.2. Deptropine Induced Autophagosome Formation but Did Not Cause Degradation of the SQSTM1/p62 Autophagic Substrate
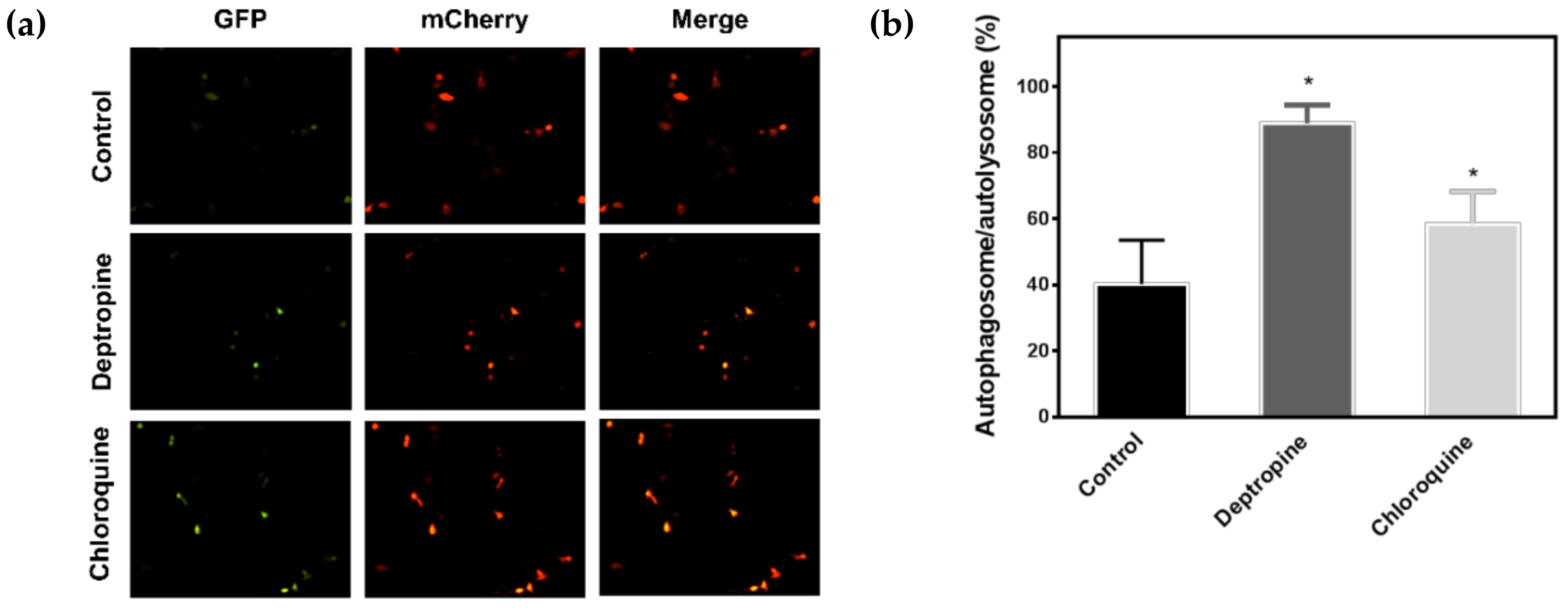
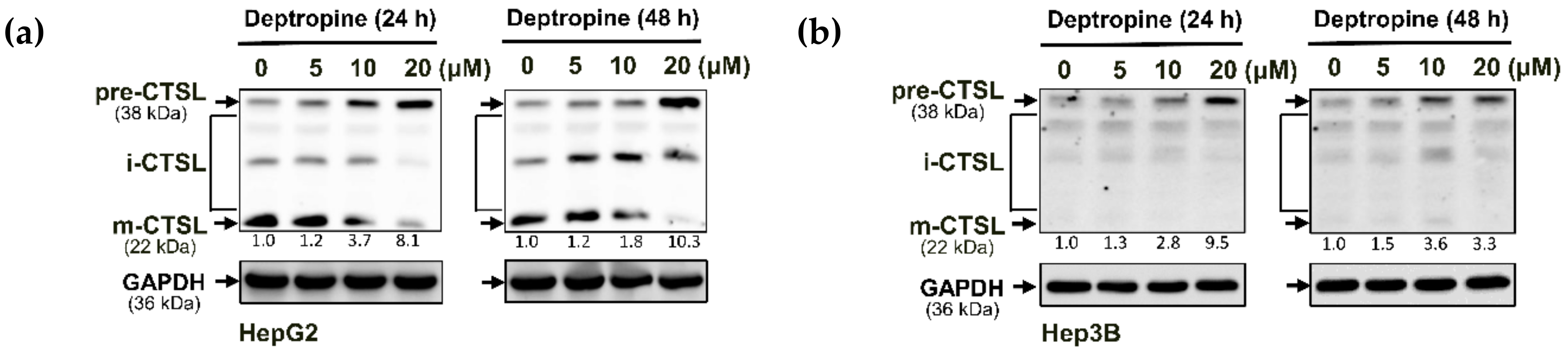
2.3. Deptropine Blocked Basal Autophagy through Blocking Autophagosome and Lysosome Fusion
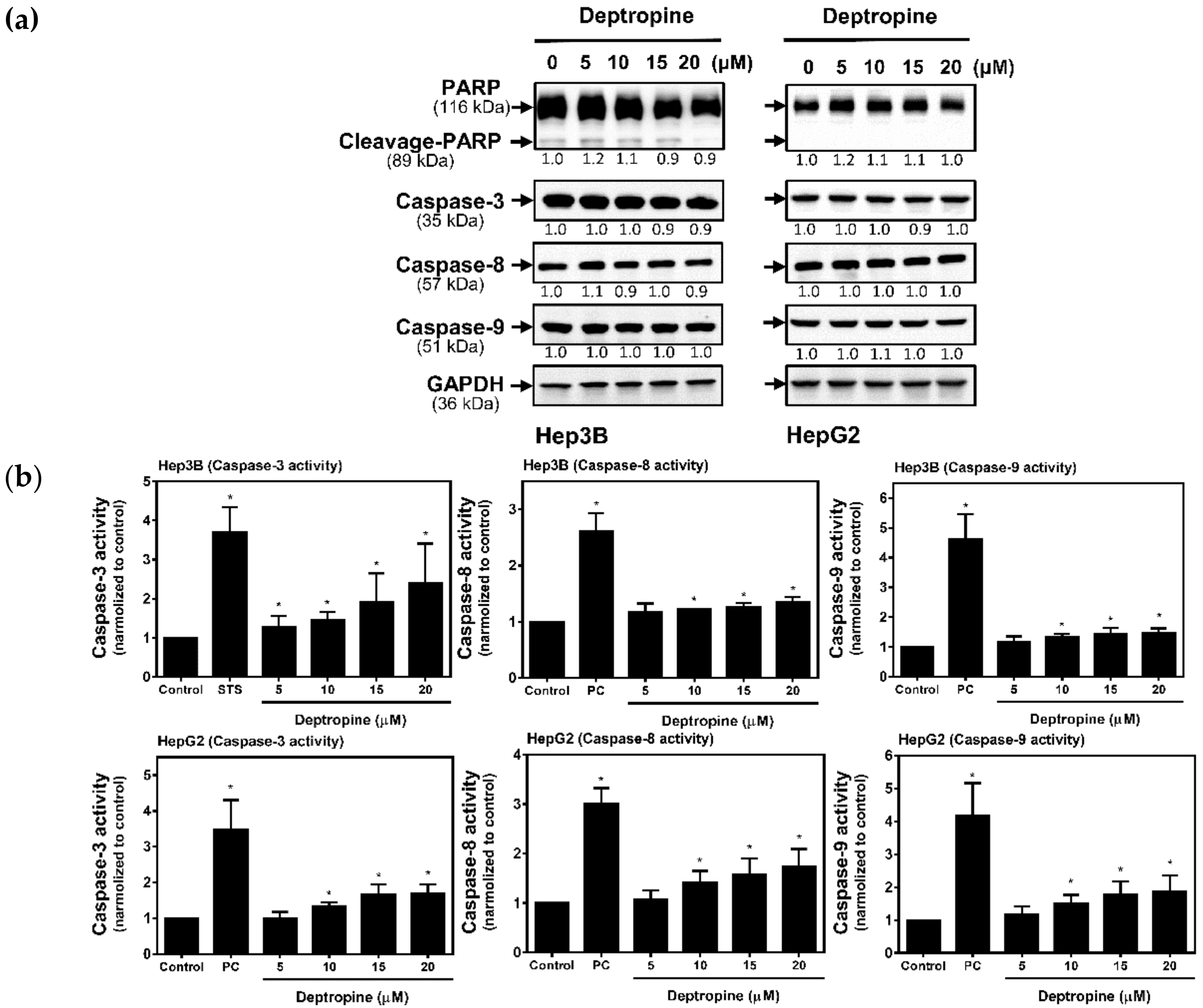
2.4. Deptropine Induced Limited Caspase Activation
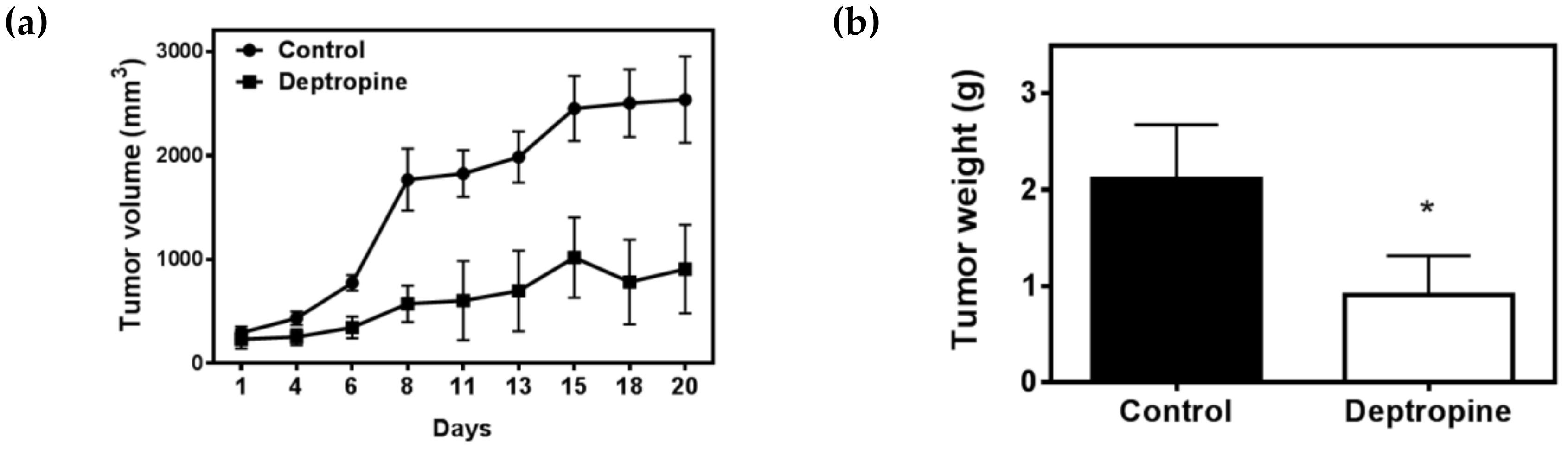
2.5. Deptropine Inhibited Hepatoma Tumor Growth in Nude Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Assay
4.4. Western Blot Analysis
4.5. Transient Transfection
4.6. Immunofluorescence Staining
4.7. Caspase Activity Assay
4.8. Antitumor Nude Mice Experiment
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, S.-N.; Wang, J.-H.; Su, C.-W.; Wang, T.-E.; Dai, C.-Y.; Chen, C.-H.; Chen, R.-C.; Yang, S.-S.; Hung, C.-F.; Huang, S.-F.; et al. Management consensus guideline for hepatocellular carcinoma: 2016 updated by the Taiwan liver cancer association and the gastroenterological society of Taiwan. J. Formos. Med. Assoc. 2018, 117, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16019. [Google Scholar] [CrossRef]
- Nault, J.-C.; Sutter, O.; Nahon, P.; Ganne-Carrié, N.; Seror, O. Percutaneous treatment of hepatocellular carcinoma: State of the art and innovations. J. Hepatol. 2018, 68, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-M.; Feng, C.-W.; Chen, S.C.-C.; Hsu, C.-D. Unexpected remission of hepatocellular carcinoma (HCC) with lung metastasis to the combination therapy of thalidomide and cyproheptadine: Report of two cases and a preliminary HCC cell line study. BMJ Case Rep. 2012, 2012, 2012007180. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-M.; Feng, C.-W.; Lu, C.-L.; Lee, M.-Y.; Chen, C.-Y.; Chen, S.C.-C. Cyproheptadine significantly improves the overall and progression-free survival of sorafenib-treated advanced HCC patients. Jpn. J. Clin. Oncol. 2015, 45, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.-C.; Lee, W.-H.; Wu, A.T.; Chow, J.-M.; Chang, C.-L.; Yuan, K.S.-P.; Wu, S.-Y. Cyproheptadine use in hepatocellular carcinoma. Am. J. Cancer Res. 2017, 7, 584–602. [Google Scholar]
- Feng, Y.-M.; Feng, C.-W.; Chen, S.-Y.; Hsieh, H.-Y.; Chen, Y.-H.; Hsu, C.-D. Cyproheptadine, an antihistaminic drug, inhibits proliferation of hepatocellular carcinoma cells by blocking cell cycle progression through the activation of P38 MAP kinase. BMC Cancer 2015, 15, 134. [Google Scholar] [CrossRef] [Green Version]
- Chávez-López, M.D.G.; Pérez-Carreón, J.I.; Zúñiga-García, V.; Díaz-Chávez, J.; Herrera, L.A.; Caro-Sánchez, C.H.; Acuña-Macías, I.; Gariglio, P.; Hernández-Gallegos, E.; Chiliquinga, A.J.; et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 2015, 36, 6149–6158. [Google Scholar] [CrossRef]
- Döbbeling, U.; Waeckerle-Men, Y.; Zabel, F.; Graf, N.; Kündig, T.M.; Johansen, P. The antihistamines clemastine and desloratadine inhibit STAT3 and c-Myc activities and induce apoptosis in cutaneous T-cell lymphoma cell lines. Exp. Dermatol. 2013, 22, 119–124. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, M.K.; Kim, S.Y.; Lee, C.H. Novel suppressive effects of ketotifen on migration and invasion of MDA-MB-231 and HT-1080 cancer cells. Biomol. Ther. 2014, 22, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soule, B.P.; Simone, N.L.; DeGraff, W.G.; Choudhuri, R.; Cook, J.A.; Mitchell, J.B. Loratadine dysregulates cell cycle progression and enhances the effect of radiation in human tumor cell lines. Radiat. Oncol. 2010, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.-Y.; Cheng, C.-L.; Ho, H.-C.; Wang, S.-S.; Chiu, K.-Y.; Su, C.-K.; Ou, Y.-C.; Lin, C.-C. Nortriptyline induces mitochondria and death receptor-mediated apoptosis in bladder cancer cells and inhibits bladder tumor growth in vivo. Eur. J. Pharmacol. 2015, 761, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2010, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Towers, C.G.; Thorburn, A. Therapeutic targeting of autophagy. EBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.G.; Jeon, T. Modulation of the autophagy-lysosomal pathway in hepatocellular carcinoma using small molecules. Molecules 2020, 25, 1580. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Deng, W.; Zhang, S.; Cai, N.; Jiao, S.; Song, J.; Wei, L. Paradoxical roles of autophagy in different stages of tumorigenesis: Protector for normal or cancer cells. Cell Biosci. 2013, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2019, 27, 843–857. [Google Scholar] [CrossRef]
- Lu, Y.; Dong, S.; Hao, H.B.; Li, C.; Zhu, K.; Guo, W.; Wang, Q.; Cheung, K.-H.; Wong, C.W.; Wu, W.-T.; et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy 2014, 10, 1895–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, G.-H.; Xie, X.; Xu, F.; Shi, X.; Wang, Y.; Deng, L. Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology 2014, 67, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Hou, Y.-F.; Melan, M.; Chen, X.; Stolz, N.B.; Shao, Z.-M.; Yin, X.-M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2006, 282, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Criado, P.R.; Criado, R.F.J.; Maruta, C.W.; Filho, C.D.M. Histamine, histamine receptors and antihistamines: New concepts. An. Bras. Dermatol. 2010, 85, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Faustino, A.; Ferreira, R.; Gama, A.; Oliveira, P.A.; Ginja, M. Antihistamines as promising drugs in cancer therapy. Life Sci. 2017, 172, 27–41. [Google Scholar] [CrossRef]
- Massari, N.A.; Nicoud, M.B.; Medina, V.A. Histamine receptors and cancer pharmacology: An update. Br. J. Pharmacol. 2018, 177, 516–538. [Google Scholar] [CrossRef] [Green Version]
- Tozzi, S.; Roth, F.E.; Tabachnick, I.I.A. The pharmacology of azatadine, a potential antiallergy drug. Inflamm. Res. 1974, 4, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Jaffer, T.; Eguchi, S.; Wang, Z.; Linkermann, A.; Ma, D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015, 6, e1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, A.P. Anoikis. Cell Death Differ. 2005, 12, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Suk, F.-M.; Chang, C.-C.; Lin, R.-J.; Lin, S.-Y.; Chen, Y.-T.; Liang, Y.-C. MCPIP3 as a potential metastasis suppressor gene in human colorectal cancer. Int. J. Mol. Sci. 2018, 19, 1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, F.-M.; Chang, C.-C.; Lin, R.-J.; Lin, S.-Y.; Liu, S.-C.; Jau, C.-F.; Liang, Y.-C. ZFP36L1 and ZFP36L2 inhibit cell proliferation in a cyclin D-dependent and p53-independent manner. Sci. Rep. 2018, 8, 2742. [Google Scholar] [CrossRef] [Green Version]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Sci. 2018, 359, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-J.; Chu, J.-S.; Chien, H.-L.; Tseng, C.-H.; Ko, P.-C.; Mei, Y.-Y.; Tang, W.-C.; Kao, Y.-T.; Cheng, H.-Y.; Liang, Y.-C.; et al. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J. Immunol. 2014, 193, 4159–4168. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-D.; Lin, S.-Y.; Ho, Y.-S.; Pan, S.; Hung, L.-F.; Tsai, S.-H.; Lin, J.-K.; Liang, Y.-C. Involvement of C-jun N-terminal kinase activation in 15-deoxy-delta 12,14-prostaglandin J2-and prostaglandin A1-induced apoptosis in AGS gastric epithelial cells. Mol. Carcinog. 2003, 37, 16–24. [Google Scholar] [CrossRef]
- Liu, J.-J.; Wu, H.-H.; Chen, T.-H.; Leung, W.; Liang, Y.-C. 15,16-Dihydrotanshinone I from the functional food salvia miltiorrhiza exhibits anticancer activity in human HL-60 leukemia cells: In vitro and in vivo studies. Int. J. Mol. Sci. 2015, 16, 19387–19400. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug. | Hep3B IC50 (μM) | HepG2 IC50 (μM) |
|---|---|---|
| Azatadine | >50.00 | >50.00 |
| Cyproheptadine | 23.00 ± 0.28 | 29.39 ± 0.58 |
| Deptropine | 9.98 ± 0.12 | 9.75 ± 0.11 |
| Ketotifen | 18.46 ± 0.11 | 23.85 ± 0.18 |
| Pizotifen | 31.73 ± 0.21 | 31.69 ± 0.16 |
| Rupatadine | 13.35 ± 0.14 | 14.68 ± 0.12 |
| Desloratadine | 21.59 ± 0.20 | 32.09 ± 0.64 |
| Loratadine | 18.27 ± 0.22 | 21.46 ± 0.19 |
| Cyclobenzaprine | 23.98 ± 0.33 | 26.79 ± 0.45 |
| Amineptine | >50.00 | >50.00 |
| Amitriptyline | 21.00 ± 0.20 | 15.15 ± 0.21 |
| Nortriptyline | 13.22 ± 0.27 | 17.10 ± 0.21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.-C.; Chang, C.-C.; Sheu, M.-T.; Lin, S.-Y.; Chung, C.-C.; Teng, C.-T.; Suk, F.-M. The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion. Cancers 2020, 12, 1610. https://doi.org/10.3390/cancers12061610
Liang Y-C, Chang C-C, Sheu M-T, Lin S-Y, Chung C-C, Teng C-T, Suk F-M. The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion. Cancers. 2020; 12(6):1610. https://doi.org/10.3390/cancers12061610
Chicago/Turabian StyleLiang, Yu-Chih, Chi-Ching Chang, Ming-Thau Sheu, Shyr-Yi Lin, Chia-Chen Chung, Chang-Ting Teng, and Fat-Moon Suk. 2020. "The Antihistamine Deptropine Induces Hepatoma Cell Death through Blocking Autophagosome-Lysosome Fusion" Cancers 12, no. 6: 1610. https://doi.org/10.3390/cancers12061610