Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment

 , ,
, ,  , , ,
, , ,
Abstract
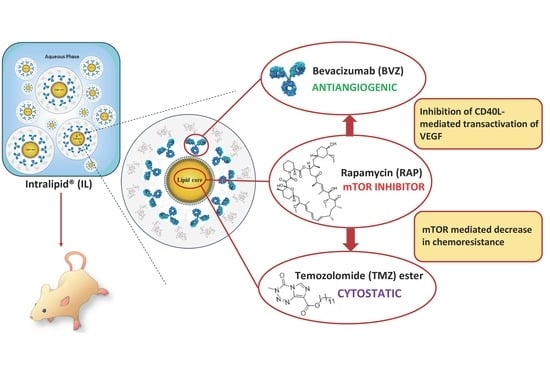
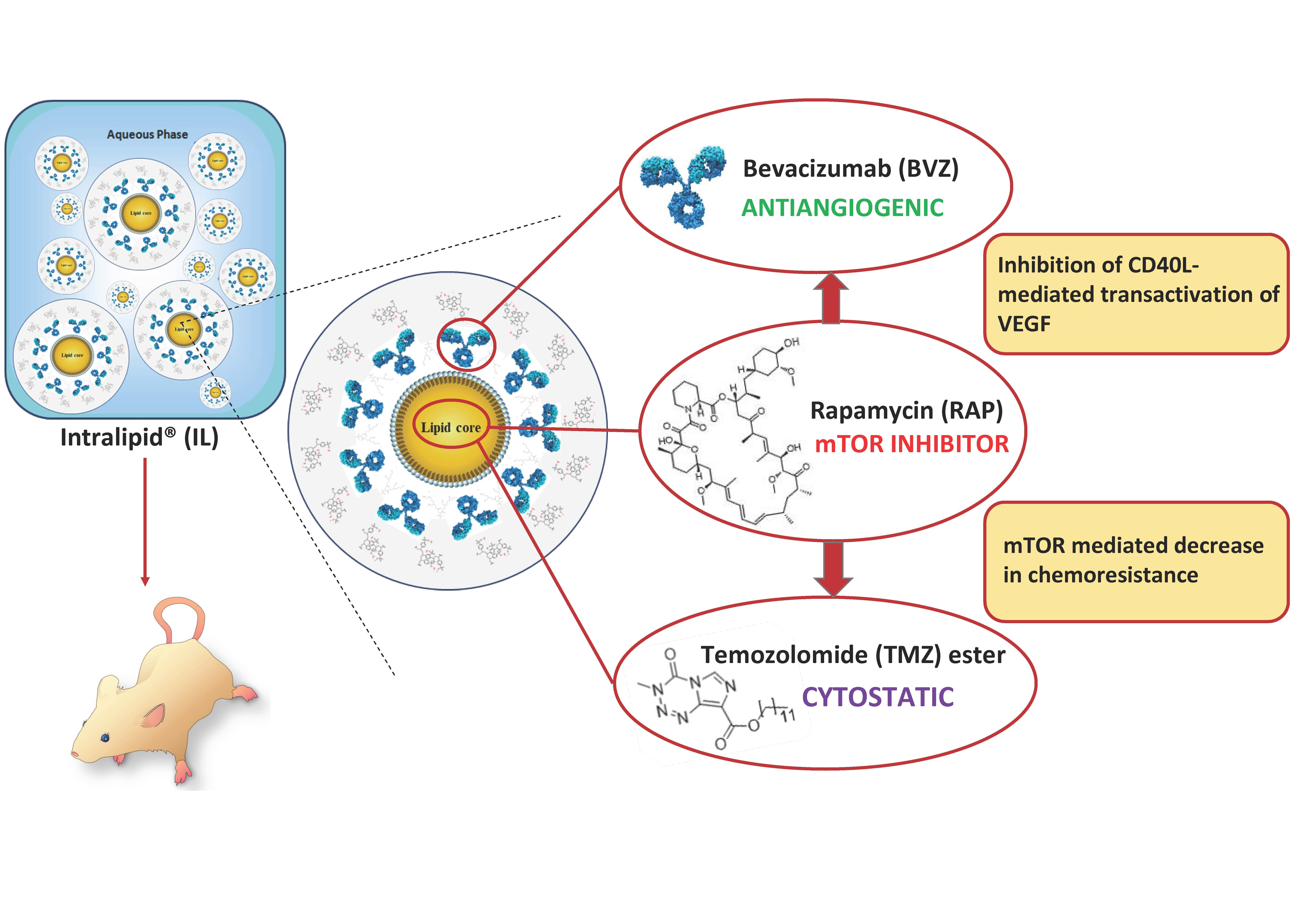
:
1. Introduction
2. Results
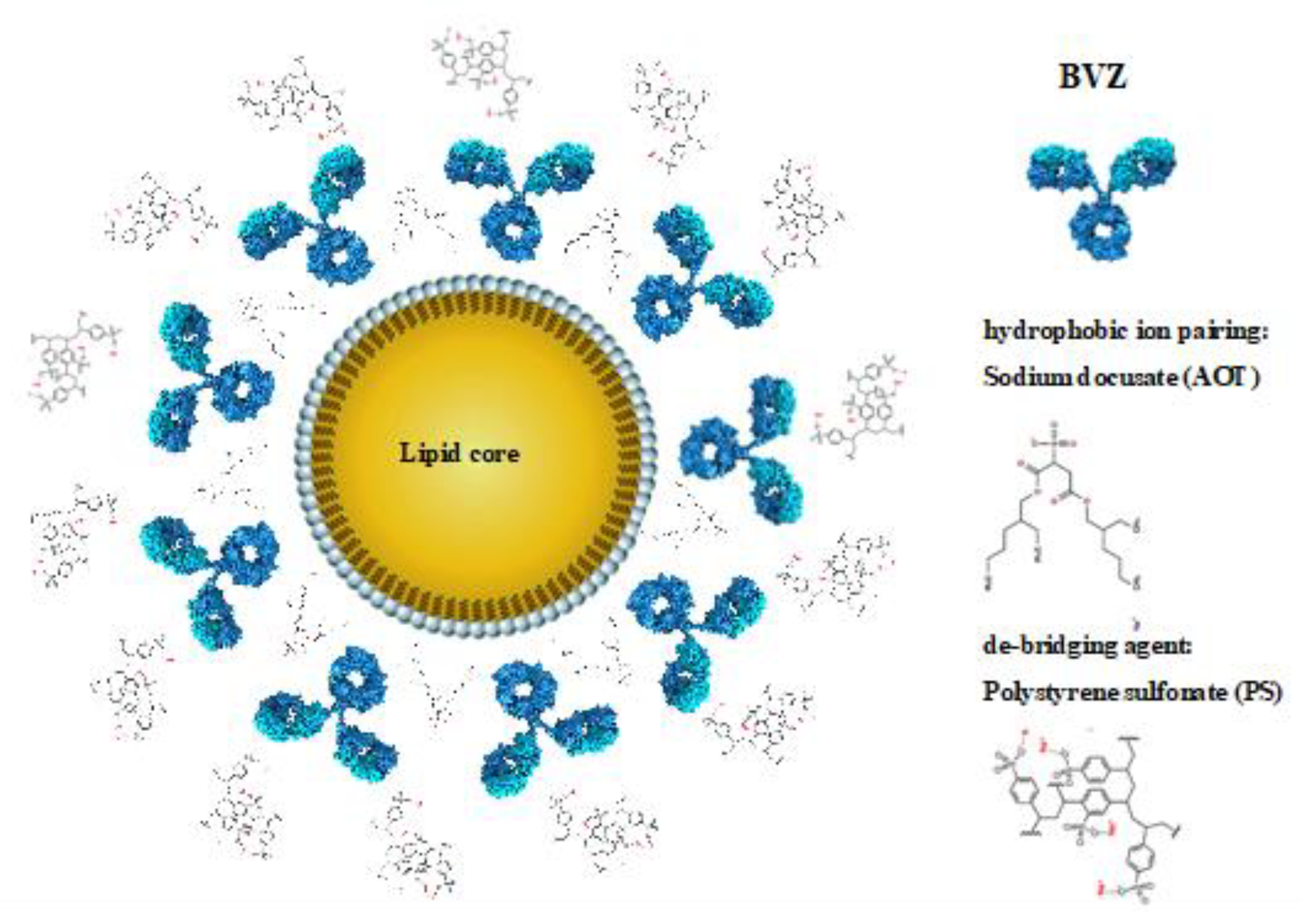
2.1. Biopharmaceutical Characterization of IL MIX
2.2. Pharmacological Characterization of IL MIX: In Vitro Studies
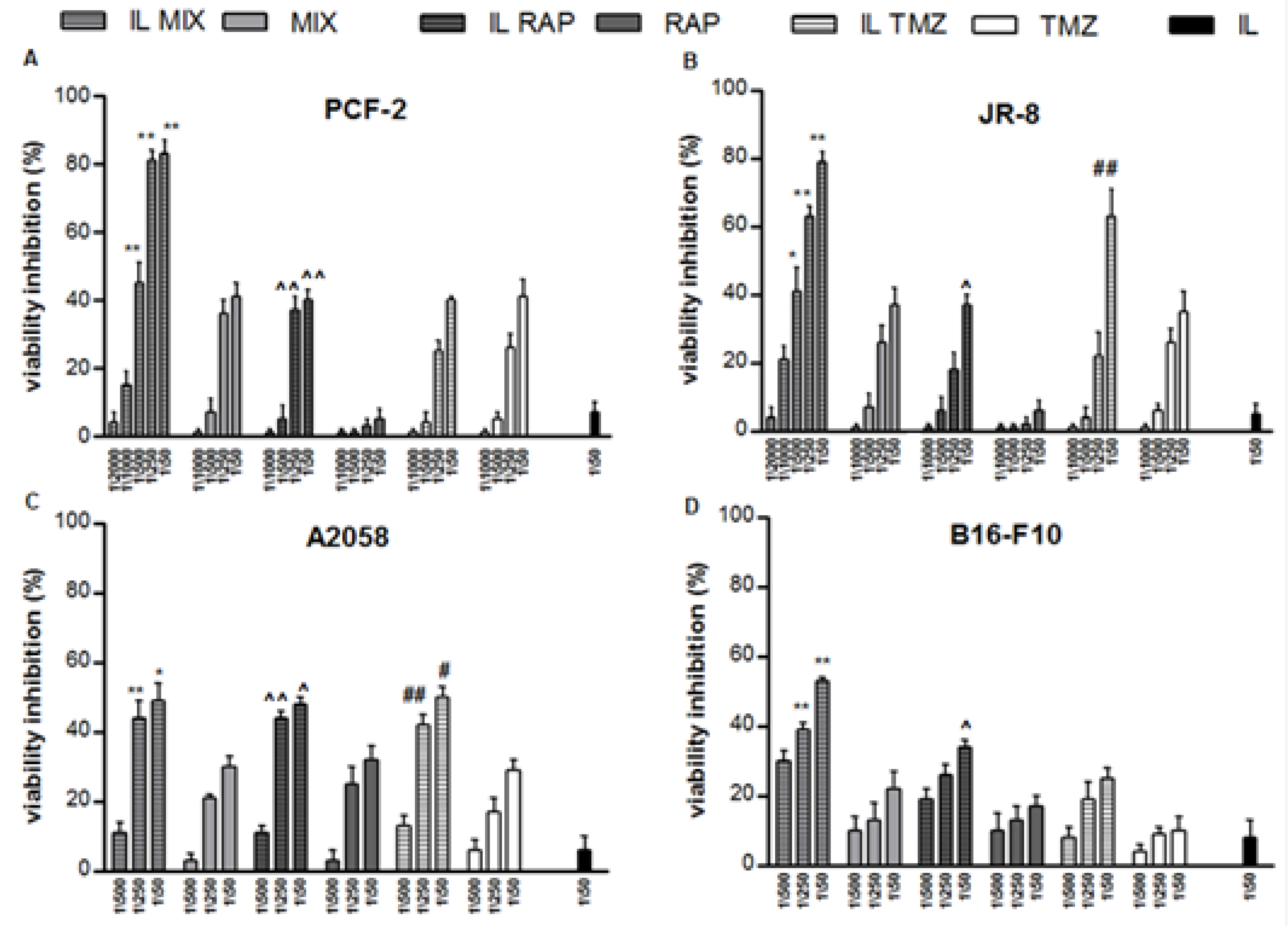
2.2.1. Inhibition of Cell Growth
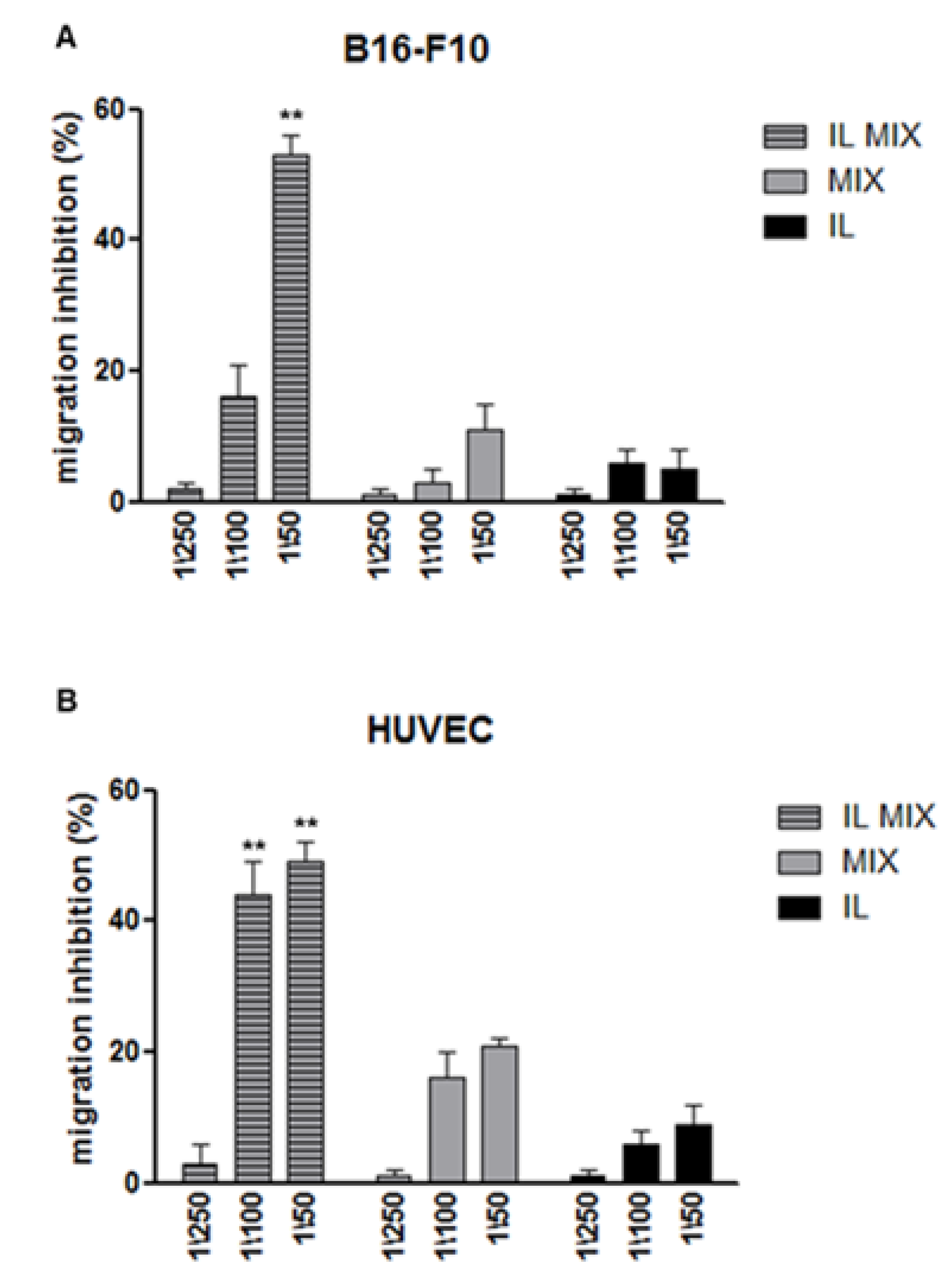
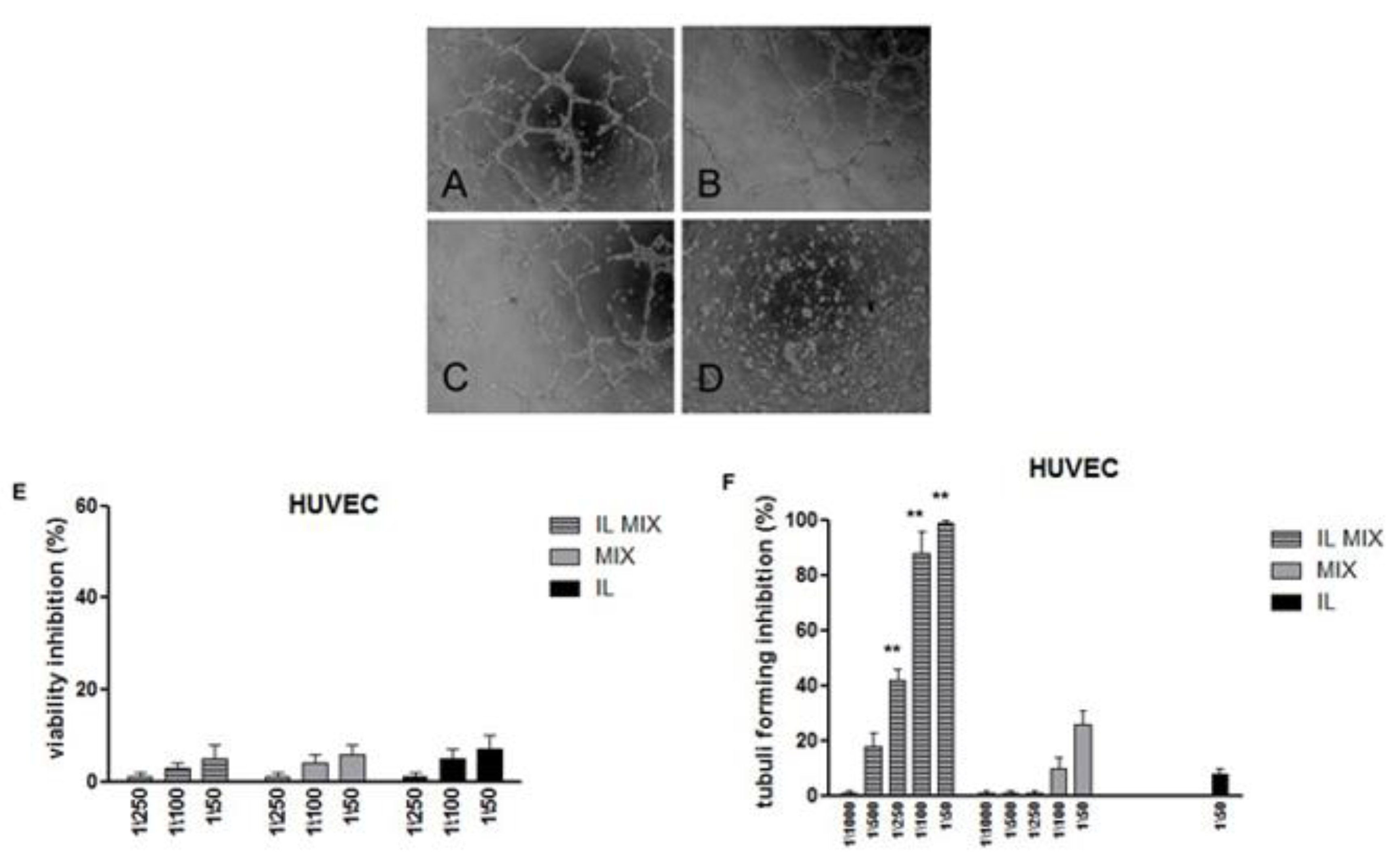
2.2.2. Inhibition of Cell Migration and Angiogenesis
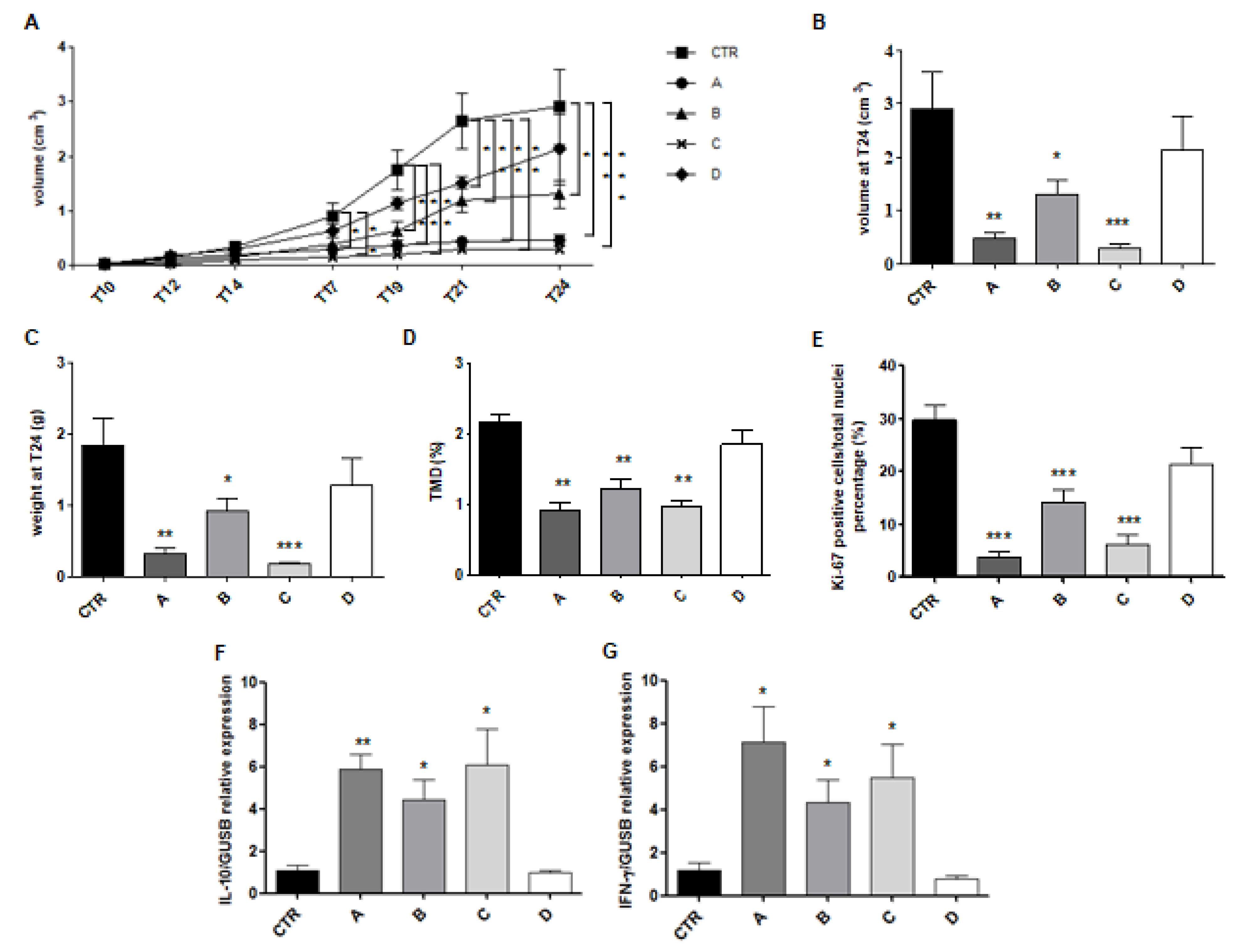
2.3. Pharmacological Characterization of IL MIX: In Vivo Studies
3. Discussion
3.1. Technological Aspects
3.2. Pharmacological Aspects
3.3. Innovation and Future Perspectives
4. Materials and Methods
4.1. Nanoemulsions
4.1.1. Formulation
4.1.2. Characterization
4.2. Cell Studies
4.2.1. Internalization
4.2.2. Cytotoxicity: WST-1 Assay
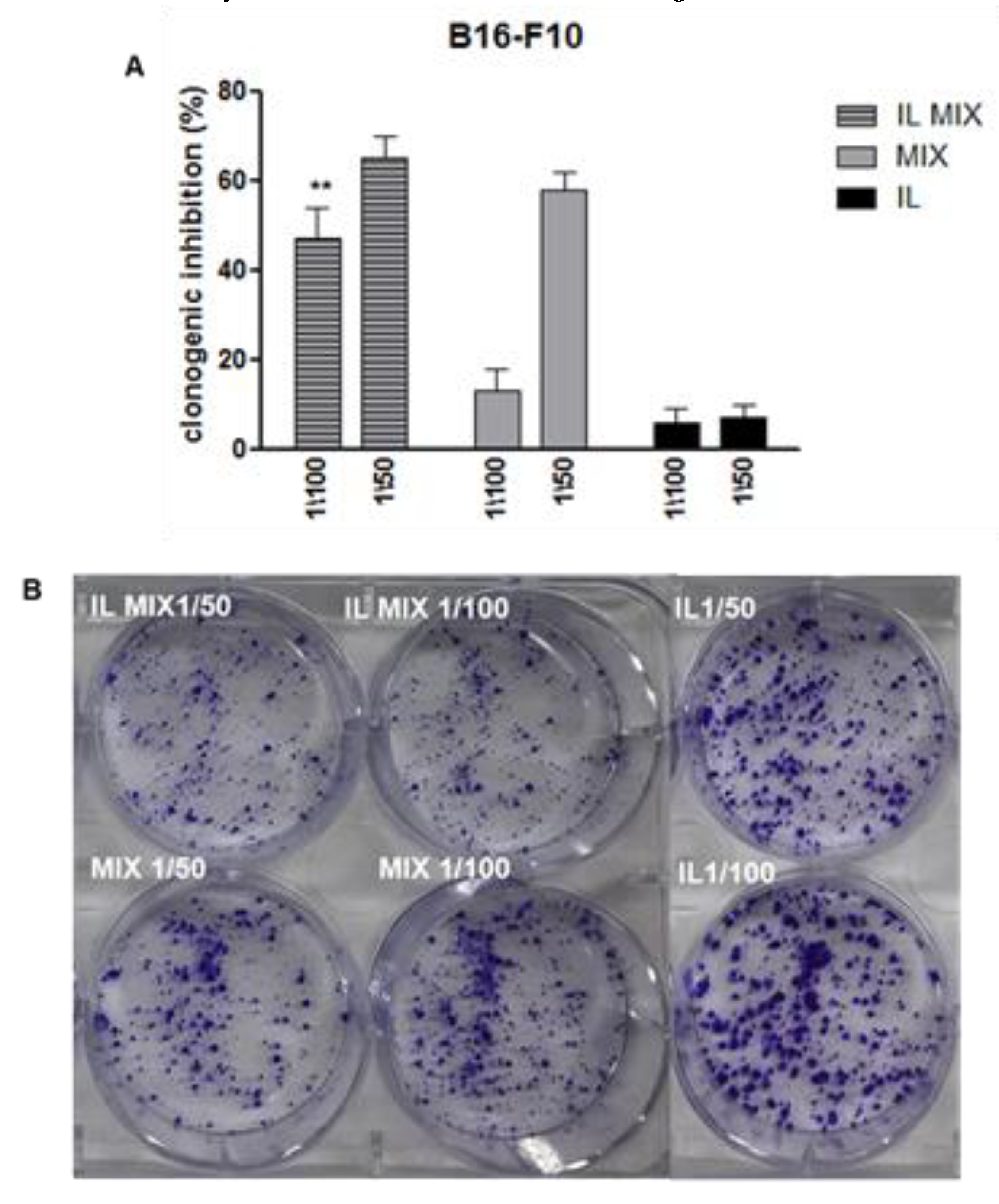
4.2.3. Proliferation: Clonogenic Assay
4.2.4. Migration: Boyden Chamber Assay
4.2.5. Angiogenesis: Tube Formation Assay
4.3. Animal Experiments
4.3.1. Immunohistochemistry on Tumour Specimens
4.3.2. Real Time PCR on Tumours
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atkinson, V. Recent advances in malignant melanoma. Intern. Med. J. 2017, 47, 1114–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PDQ Adult Treatment Editorial Board. Melanoma Treatment (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Calmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Eng. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Daga, M.; Dianzani, C.; Ferrara, B.; Nardozza, V.; Cavalli, R.; Barrera, G.; Pizzimenti, S. Latest News on Nanotechnology for Melanoma Therapy and Diagnosis. SM J. Nanotechnol. Nanomed. 2016, 2, 1002. [Google Scholar]
- Pizzimenti, S.; Dianzani, C.; Zara, G.P.; Barrera, G. Challenges and Opportunities of Nanoparticle-based Theranostics in Skin Cancer. In Nanoscience in Dermatology; Hamblin, M.R., Avci, P., Prow, T.W., Eds.; Elsevier: London, UK, 2016; pp. 177–188. [Google Scholar] [CrossRef]
- Hippalgaonkar, K.; Majumdar, S.; Kansara, V. Injectable Lipid Emulsions-Advancements, Opportunities and Challenges. AAPS Pharm. Sci. Technol. 2010, 11, 1526–1540. [Google Scholar] [CrossRef] [Green Version]
- Clemente, N.; Ferrara, B.; Luca, C.; Boggio, E.; Capucchio, M.T.; Biasibetti, E.; Schiffer, D.; Mellai, M.; Annovazzi, L.; Cangemi, L.; et al. Solid Lipid Nanoparticles Carrying Temozolomide for Melanoma Treatment. Preliminary In Vitro and In Vivo Studies. Int. J. Mol. Sci. 2018, 19, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dronca, R.S.; Allred, J.B.; Perez, D.G.; Nevala, W.K.; Lieser, E.A.; Thompson, M.; Maples, W.J.; Creagan, E.T.; Pockaj, B.A.; Kaur, J.S.; et al. Phase II Study of Temozolomide (TMZ) and Everolimus (RAD001) Therapy for Metastatic Melanoma: A North Central Cancer Treatment Group Study, N0675. Am. J. Clin. Oncol. 2014, 37, 369. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR Signaling Sensitizes Melanoma Cells to Cisplatin and Temozolomide. J. Investig. Dermatol. 2009, 129, 1500–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormond, O.; Contreras, A.G.; Meijer, E.; Datta, D.; Flynn, E.; Pal, S.; Briscoe, D.M. CD40-Induced Signaling in Human Endothelial Cells Results in mTORC2- and Akt-Dependent Expression of Vascular Endothelial Growth Factor In Vitro and In Vivo. J. Immunol. 2008, 181, 8088–8095. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Peyton, J.D.; Thompson, D.S.; Lane, C.M.; Clark, B.L.; Rubin, M.S.; Trent, D.F.; Burris, H.A., III. Bevacizumab and Everolimus in the Treatment of Patients with Metastatic Melanoma. Cancer 2010, 116, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Gallarate, M.; Peira, E.; Chirio, D.; Solazzi, I.; Giordano, S.M.; Gigliotti, C.L.; Riganti, C.; Dianzani, C. Bevacizumab loaded solid lipid nanoparticles prepared by the coacervation technique: Preliminary in vitro studies. Nanotechnology. 2015, 26, 255102. [Google Scholar] [CrossRef] [Green Version]
- Finke, J.H.; Richter, C.; Gothsch, T.; Kwade, A.; Büttgenbach, S.; Müller-Goymann, C.C. Coumarin 6 as a fluorescent model drug: How to identify properties of lipid colloidal drug delivery systems via fluorescence spectroscopy? Eur. J. Lipid Sci. Technol. 2014, 116, 1234–1246. [Google Scholar] [CrossRef]
- Pretor, S.; Bartels, J.; Lorenz, T.; Dahl, K.; Finke, J.H.; Peterat, G.; Krull, R.; Al-Halhouli, A.T.; Dietzel, A.; Buttgenbach, S.; et al. Cellular Uptake of Coumarin-6 under Microfluidic Conditions into HCE-T Cells from Nanoscale Formulations. Mol. Pharm. 2015, 12, 34–45. [Google Scholar] [CrossRef]
- Weihrauch, D.; Shumpert, S.D.; Larson, M.E.; McVey, N.; Krolikowski, J.G.; Bamkole, O.; Riess, M.L. Intralipid Increases Nitric Oxide Release from Human Endothelial Cells During Oxidative Stress. J. Parenter. Enter. Nutr. 2020. [CrossRef]
- Vilaro, S.; Llobera, M. Uptake and metabolism of Intralipid by rat liver: An electron-microscopic study. J. Nutr. 1988, 118, 932–940. [Google Scholar] [CrossRef]
- McVey, N.; Richman, M.; Struve, J.; Baumgardt, S.; Weihrauch, D. Intravenous Lipid Emulsion Induces Endocytosis in Human Coronary Artery Endothelial Cells by a CD36/Caveolin-1 Pathway. FASEB J. 2018, 32, 1. [Google Scholar]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E) BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, V.; Le Mercier, M.; De Neve, N.; Sauvage, S.; Gras, T.; Roland, I.; Lefranc, F.; Kiss, R. Galectin-1 Knockdown Increases Sensitivity to Temozolomide in a B16F10 Mouse Metastatic Melanoma Model. J. Investig. Dermatol. 2007, 127, 2399–2410. [Google Scholar] [CrossRef]
- Jiang, G.; Sun, C.; Li, R.H.; Wei, Z.P.; Zheng, J.N.; Liu, Y.Q. Enhanced antitumor efficacy of a novel oncolytic adenovirus combined with temozolomide in the treatment of melanoma in vivo. J. Cancer Res. Clin. Oncol. 2015, 141, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Karmali, R.A.; Maxuitenko, Y.Y.; Gorman, G.S.; Qu, Z. Combinatorial treatment with carboxyamidotriazole-orotate and temozolomide in sc-implanted human LOX IMVI melanoma xenografts. J. Solid Tumors 2012, 2, 13–28. [Google Scholar] [CrossRef]
- Middleton, M.R.; Kelly, J.; Thatcher, N.; Donnelly, D.J.; McElhinney, R.S.; McMurry, T.B.; McCormick, J.E.; Margison, G.P. O6-(4-bromothenyl)guanine improves the therapeutic index of temozolomide against A375M melanoma xenografts. Int. J. Cancer 2000, 252, 248–252. [Google Scholar] [CrossRef]
- Walker, E.J.; Su, H.; Shen, F.; Degos, V.; Amend, G.; Jun, K.; Young, W.L. Bevacizumab attenuates VEGF-induced angiogenesis and vascular malformations in the adult mouse brain. Stroke 2012, 43, 1925–1930. [Google Scholar] [CrossRef] [Green Version]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Antoch, M.P.; Blagosklonny, M.V. Rapamycin Extends Maximal Lifespan in Cancer-Prone Mice. Am. J. Pathol. 2010, 176, 2092–2097. [Google Scholar] [CrossRef]
- Abdelnour-Berchtold, E.; Cerantola, Y.; Roulin, D.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Rapamycin-mediated FOXO1 Inactivation Reduces the Anticancer Efficacy of Rapamycin. Anticancer Res. 2010, 30, 799–804. [Google Scholar]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [Green Version]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamilvanan, S. Formulation of multifunctional oil-in-water nanosized emulsions for active and passive targeting of drugs to otherwise inaccessible internal organs of the human body. Int. J. Pharm. 2009, 381, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Kadam, A.; Wan, K.W.; Ahmed, W.; Taylor, K.M.; Elhissi, A.M. Novel paclitaxel formulations solubilized by parenteral nutrition nanoemulsions for application against glioma cell lines. Int. J. Pharm. 2016, 506, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, K.; Sun, J.; Liu, K.; Liu, X.; He, Z. Preparation and characterization of a submicron lipid emulsion of docetaxel: Submicron lipid emulsion of docetaxel. Drug Dev. Ind. Pharm. 2008, 34, 1227–1237. [Google Scholar] [CrossRef]
- Ganta, S.; Paxton, J.W.; Baguley, B.C.; Garg, S. Pharmacokinetics and pharmacodynamics of chlorambucil delivered in parenteral emulsion. Int. J. Pharm. 2008, 360, 115–121. [Google Scholar] [CrossRef]
- Anuchapreeda, S.; Fukumori, Y.; Okonogi, S.; Ichikawa, H. Preparation of lipid nanoemulsions incorporating curcumin for cancer therapy. J. Nanotechnol. 2011, 2012. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Chen, H.; Lin, X.; Tang, X. Pharmacokinetics, tissue distribution and safety of cinnarizine delivered in lipid emulsion. Int. J. Pharm. 2010, 383, 264–270. [Google Scholar] [CrossRef]
- Shi, K.; Xia, Y.; Wang, Q.; Wu, Y.; Dong, X.; Chen, C.; Tang, W.; Zhang, Y.; Luo, M.; Wang, X.; et al. The effect of lipid emulsion on pharmacokinetics and tissue distribution of bupivacaine in rats. Anesthesia Analg. 2013, 116, 804–809. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, A.; Shibayama, Y.; Mizota, A.; Yasuno, A.; Ikeda, M.; Sasagawa, K.; Kobayashi, T.; Hisaoka, M. Lipid emulsions of palmitoylrhizoxin: Effects of composition on lipolysis and biodistribution. Biopharm Drug Dispos. 1996, 17, 331–342. [Google Scholar] [CrossRef]
- Buysa, M.; Scheepersa, P.A.; Levina, A.I. Lipid emulsion therapy: Non-nutritive uses of lipid emulsions in anaesthesia and intensive care. S. Afr. J. Anaesth. Analg. 2015, 21, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Stawny, M.; Olijarczyk, R.; Jaroszkiewicz, E.; Jelińska, A. Pharmaceutical point of view on parenteral nutrition. Sci. World J. 2013, 2013, 415310. [Google Scholar] [CrossRef] [Green Version]
- Annovazzi, L.; Schiffer, D.; Mellai, M.; Marina, G.; Battaglia, L.; Chirio, D.; Peira, E.; Muntoni, E.; Chegaev, K.; Barge, A.; et al. Solid Lipid Nanoparticles Loaded with Antitumor Lipophilic Prodrugs Aimed to Glioblastoma Treatment: Preliminary Studies on Cultured Cells. J. Nanosci. Nanotechnol. 2017, 17, 3606–3614. [Google Scholar] [CrossRef]
- Suppasansatorn, P.; Wang, G.; Conway, B.R.; Wang, W.; Wang, Y. Skin delivery potency and antitumor activities of temozolomide ester prodrugs. Cancer Lett. 2006, 244, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Navas, N.; Herrera, A.; Martínez-Ortega, A.; Salmerón-García, A.; Cabeza, J.; Cuadros-Rodríguez, L. Quantification of an intact monoclonal antibody, rituximab, by (RP)HPLC/DAD in compliance with ICH guidelines. Anal. Bioanal. Chem. 2013, 405, 9351–9363. [Google Scholar] [CrossRef] [PubMed]
- Mortz, E.; Krogh, T.N.; Vorum, H.; Görg, A. Improved silver staining protocols for high sensitivity protein identification using matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics 2001, 1, 1359–1363. [Google Scholar] [CrossRef]
- Tadjiki, S.; Moldenhauer, E.; Klein, T. Study of Temperature Stability, Fragmentation and Aggregation of Recombinant Antibodies with Asymmetrical Flow FFF and UV. Postnova Technical Document ID0013. Available online: https://www.postnova.com/images/pdf/Postnova_ID0013_AN_AntibodiesAF4andUV.pdf (accessed on 9 May 2020).
- Palais, C.; Capelle, M.; Arvinte, T. Studies of Loose Protein Aggregates by Flow Field-Flow Fractionation (FFF) Coupled to Multi-Angle Laser Light Scattering (MALLS). In Field-Flow Fractionation in Biopolymer Analysis; Williams, S.K.R., Caldwell, K.D., Eds.; Springer: Vienna, Austria, 2012; pp. 103–112. [Google Scholar] [CrossRef]
- Dianzani, C.; Brucato, L.; Gallicchio, M.; Rosa, A.C.; Collino, M.; Fantozzi, R. Celecoxib modulates adhesion of HT29 colon cancer cells to vascular endothelial cells by inhibiting ICAM-1 and VCAM-1 expression. Br. J. Pharmacol. 2008, 153, 1153–1161. [Google Scholar] [CrossRef]
- Pescina, S.; Ferrari, G.; Govoni, P.; Macaluso, C.; Padula, C.; Santi, P.; Nicoli, S. In-vitro permeation of bevacizumab through human sclera: Effect of iontophoresis application. J. Pharm Pharmacol. 2010, 62, 1189–1194. [Google Scholar] [CrossRef]
- Srinivasan, R.; Tan, L.P.; Wu, H.; Yang, P.Y.; Kalesh, K.A.; Yao, S.Q. High-throughput synthesis of azide libraries suitable for direct “click” chemistry and in situ screening. Org. Biomol. Chem. 2009, 7, 1821–1828. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP rapamycin FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, X.; He, Z.; Jin, Y.; Gong, L.; Mi, A. Reduction of azides to amines or amides with zinc and ammonium chloride as reducing agent. Synth. Commun. 2002, 32, 3279–3284. [Google Scholar] [CrossRef]
- Boggio, E.; Dianzani, C.; Gigliotti, C.L.; Soluri, M.F.; Clemente, N.; Cappellano, G.; Toth, E.; Raineri, D.; Ferrara, B.; Comi, C.; et al. Thrombin Cleavage of Osteopontin Modulates Its Activities in Human Cells In Vitro and Mouse Experimental Autoimmune Encephalomyelitis In Vivo. J. Immunol. Res. 2016, 2016, 9345495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigliotti, C.L.; Minelli, R.; Cavalli, R.; Occhipinti, S.; Barrera, G.; Pizzimenti, S.; Cappellano, G.; Boggio, E.; Conti, L.; Fantozzi, R.; et al. In Vitro and In Vivo Therapeutic Evaluation of Camptothecin-Encapsulated β-Cyclodextrin Nanosponges in Prostate Cancer. J. Biomed. Nanotechnol. 2016, 12, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Passaro, C.; Borriello, F.; Vastolo, V.; Di Somma, S.; Scamardella, E.; Gigantino, V.; Franco, R.; Marone, G.; Portella, G. The oncolytic virus dl922-947 reduces IL-8/CXCL8 and MCP-1/CCL2 expression and impairs angiogenesis and macrophage infiltration inanaplastic thyroid carcinoma. Oncotarget 2016, 7, 1500–1515. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Size (nm) | 262.6 ± 15 | |
| PDI | 0.113 | |
| Zeta Potential (mV) | −33.14 ± 4.5 | |
| Recovery | RP-HPLC | TMZ 75 ± 4% RAP 100 ± 3% BVZ 64 ± 7% |
| SDS-PAGE electrophoresis/densitometry | BVZ 57 ± 8% | |
| EE% | RP-HPLC | TMZ 68 ± 4% RAP 56 ±6% |
| SDS-PAGE electrophoresis/densitometry | BVZ 47 ± 5% | |
| Formulations | iMFI (A2058 Cells) | iMFI (B16-F10 Cells) |
|---|---|---|
| Control | 501.77 | 102.82 |
| Fluorescently labelled IL | 2,613,367.19 | 1,414,784.30 |
| BVZ-FITC loaded IL | 6,193.05 | 18,342.9 |
| FITC-glycilrapamycin loaded IL | 26,891.73 | 69,574.71 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dianzani, C.; Monge, C.; Miglio, G.; Serpe, L.; Martina, K.; Cangemi, L.; Ferraris, C.; Mioletti, S.; Osella, S.; Gigliotti, C.L.; et al. Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment. Cancers 2020, 12, 1198. https://doi.org/10.3390/cancers12051198
Dianzani C, Monge C, Miglio G, Serpe L, Martina K, Cangemi L, Ferraris C, Mioletti S, Osella S, Gigliotti CL, et al. Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment. Cancers. 2020; 12(5):1198. https://doi.org/10.3390/cancers12051198
Chicago/Turabian StyleDianzani, Chiara, Chiara Monge, Gianluca Miglio, Loredana Serpe, Katia Martina, Luigi Cangemi, Chiara Ferraris, Silvia Mioletti, Sara Osella, Casimiro Luca Gigliotti, and et al. 2020. "Nanoemulsions as Delivery Systems for Poly-Chemotherapy Aiming at Melanoma Treatment" Cancers 12, no. 5: 1198. https://doi.org/10.3390/cancers12051198