Chromenopyrimidinone Controls Stemness and Malignancy by suppressing CD133 Expression in Hepatocellular Carcinoma

Abstract
:1. Introduction
2. Results
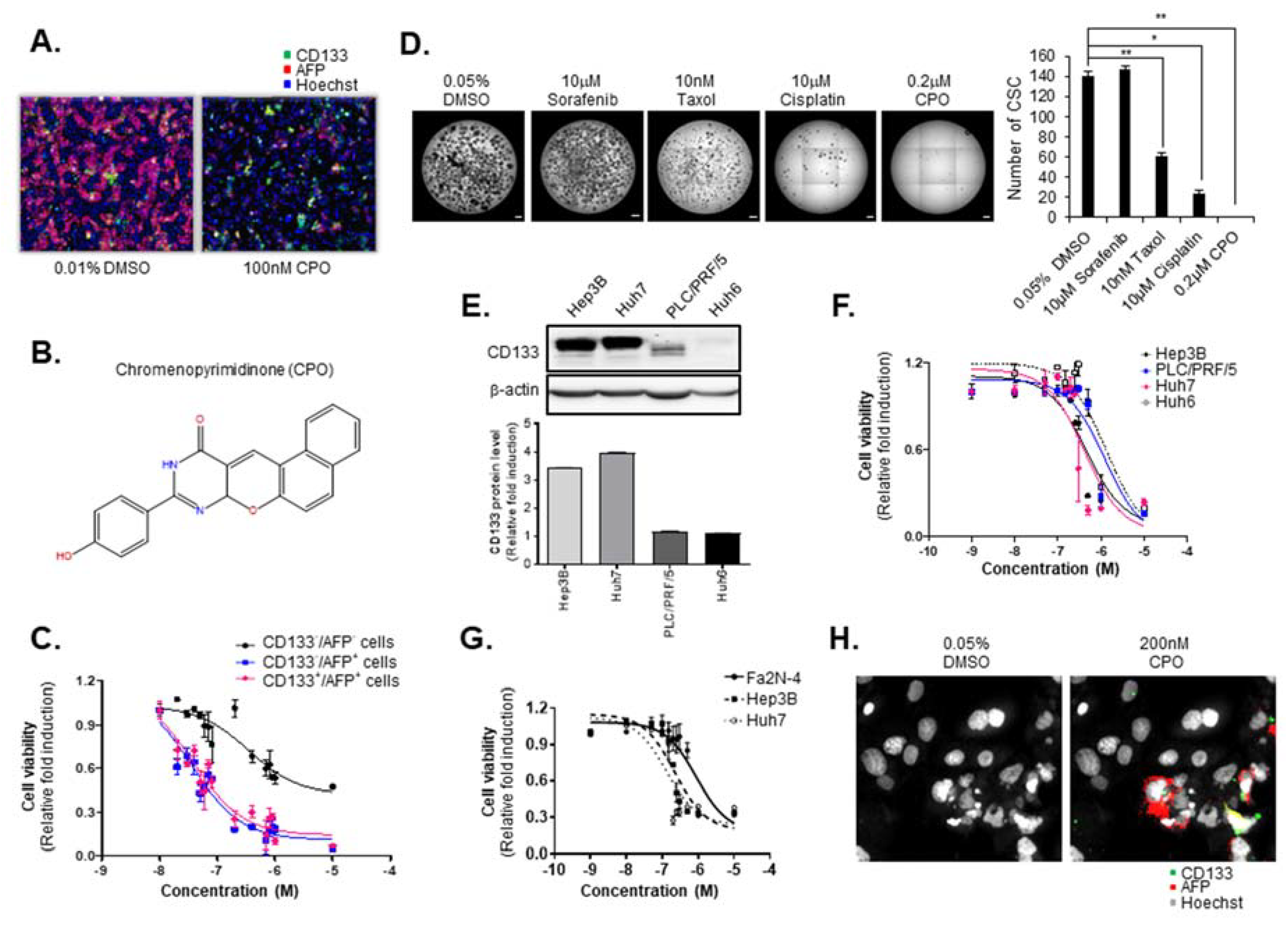
2.1. CPO Effectively Suppresses CD133+ Cells in HCC
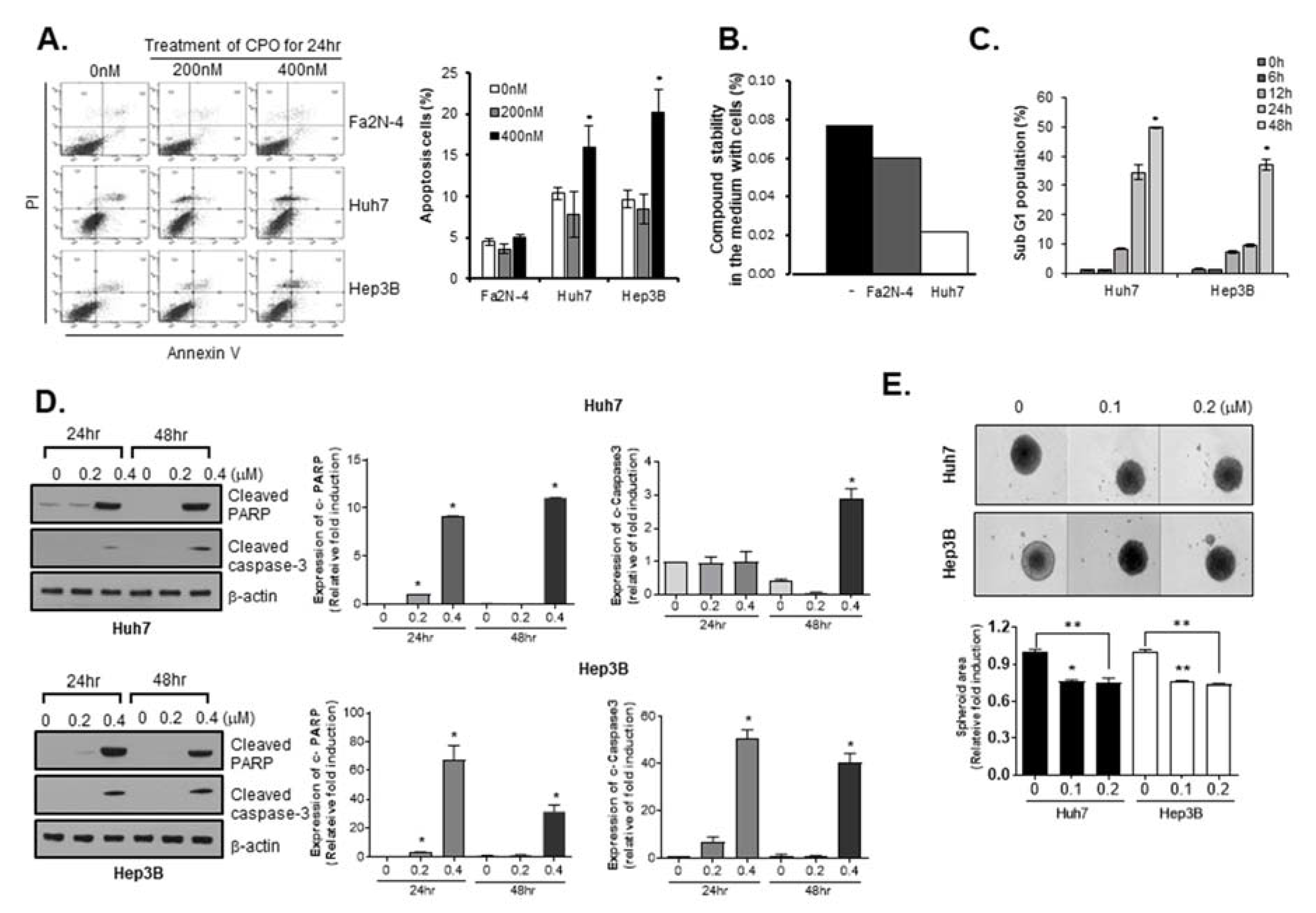
2.2. CPO Induces Apoptosis in HCC Cells
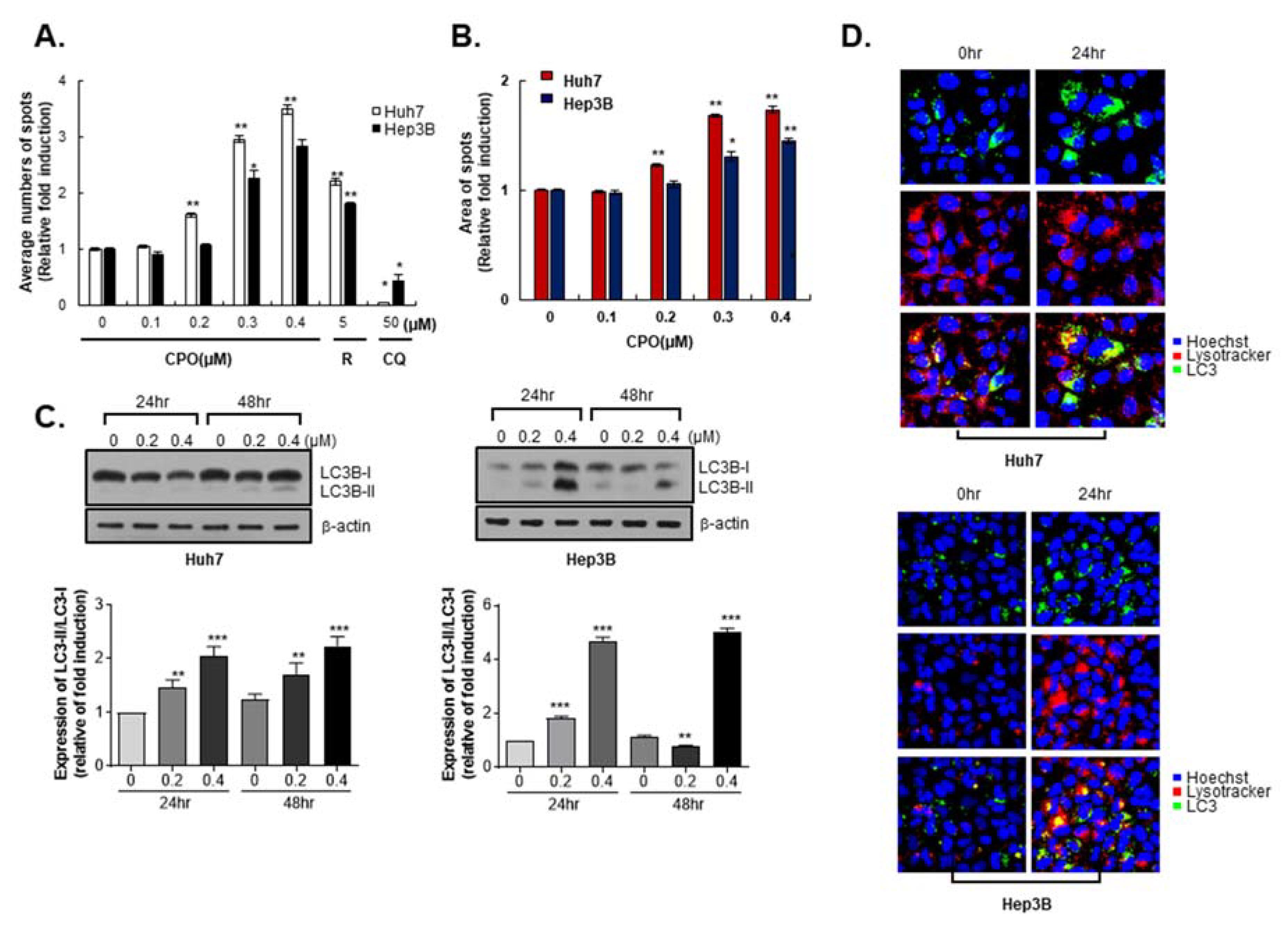
2.3. CPO Induces Autophagy in HCC Cells
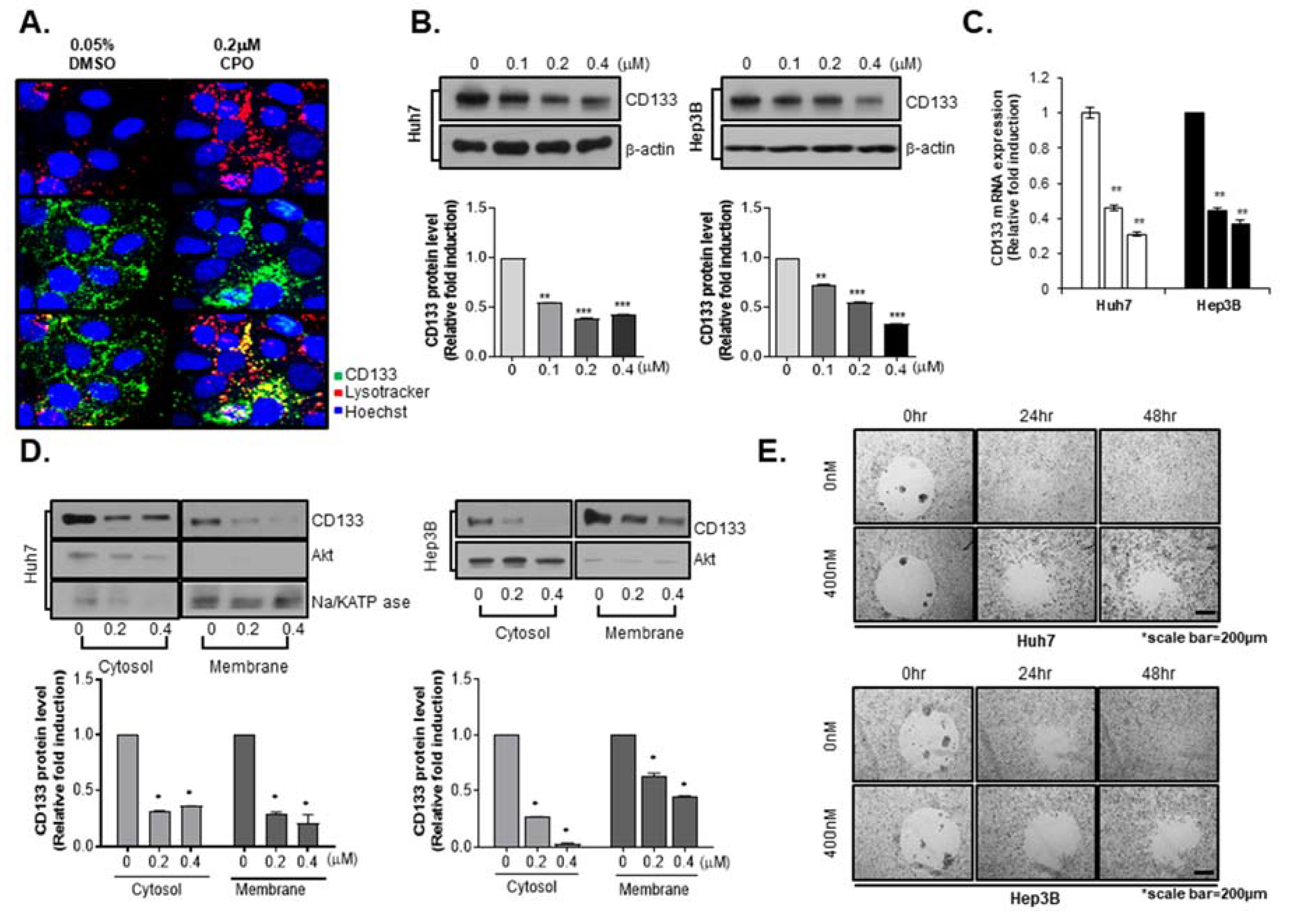
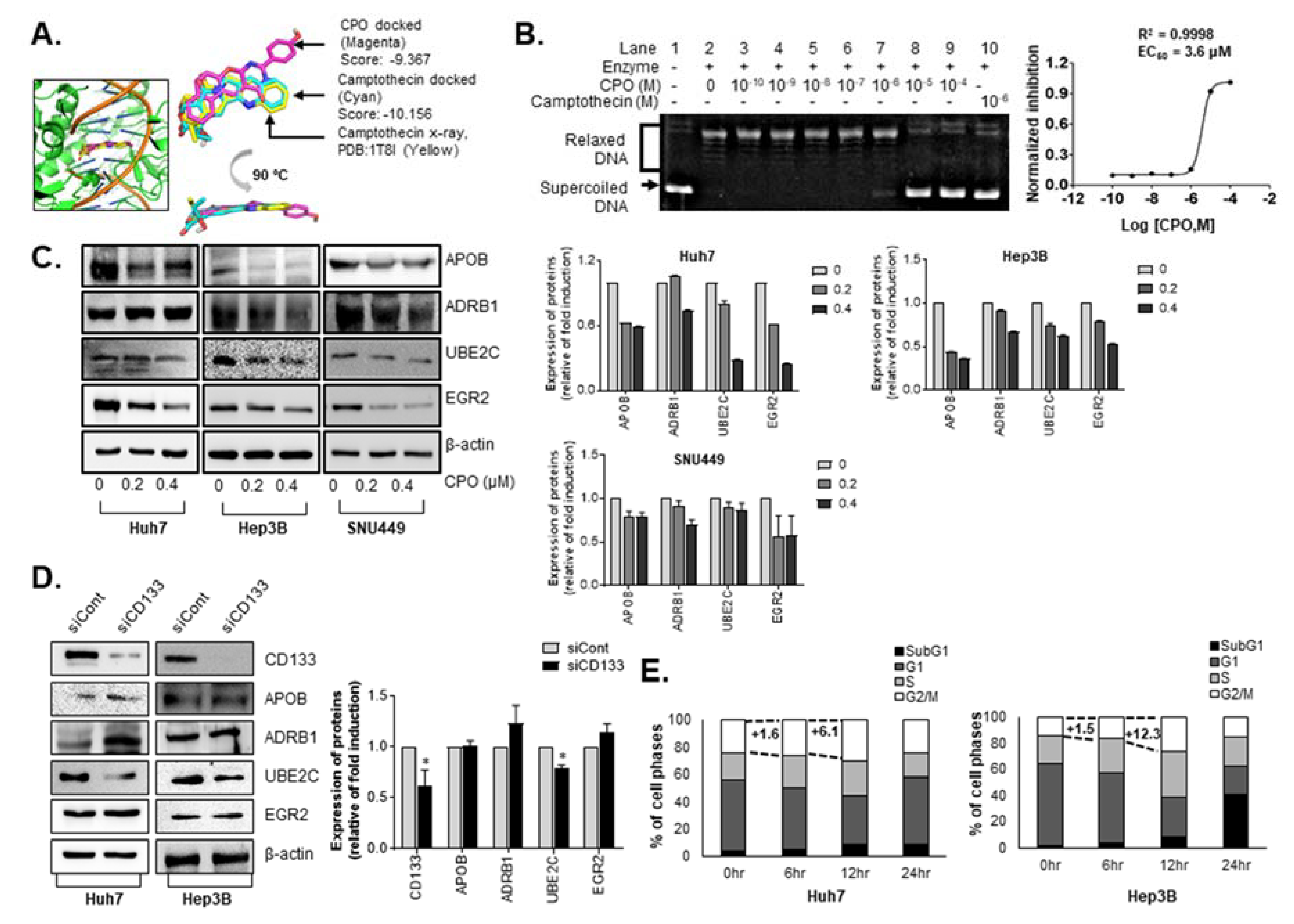
2.4. CPO Altered CD133 Subcellular Localization and Induced Degradation of CD133 in HCC
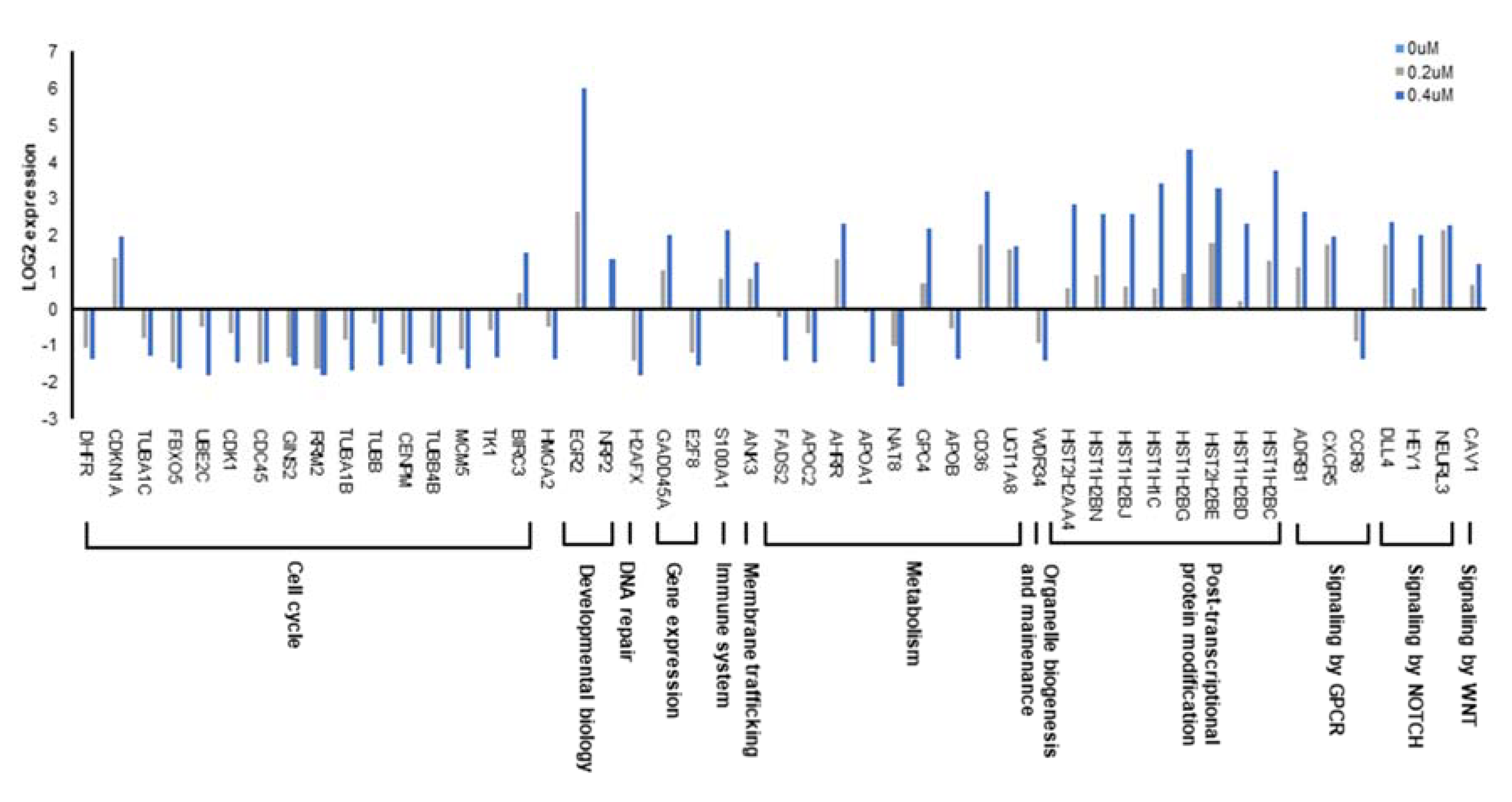
2.5. RNA-seq Profiling Reveals a Novel Target Pathway of CPO in HCC
2.6. CPO Target Cell Cycle and Lipid Metabolism Pathway in HCC
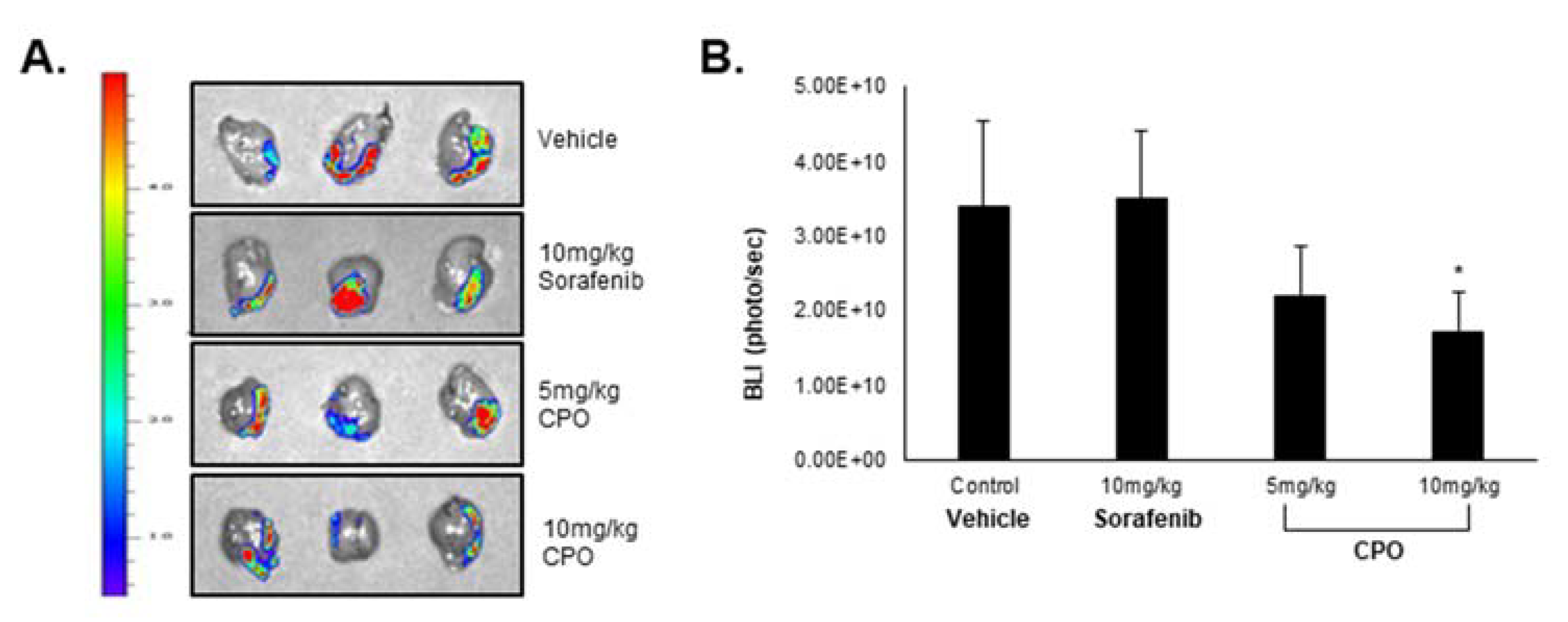
2.7. CPO Exhibits In Vivo Therapeutic Efficiency in HCC
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Mixed Culture System
4.3. Drug Sensitivity of Liver Cancer Stem Cell Spheroids
4.4. Western Blot Analysis
4.5. High Content Screening (HCS) Imaging Assay Technology
4.6. Immunocytochemistry
4.7. Apoptosis Assay
4.8. Cell Cycle Analysis
4.9. Drug Sensitivity in HCC Spheroids
4.10. Autophagy Analysis
4.11. Cell Fractionation
4.12. Cell Migration Assay
4.13. Drug Efficacy in HCC Orthotopic Mouse Model
- Group 1: Vehicle control (10% DMSO + 90% Captisol)
- Group 2: 5 mg/kg CPO
- Group 3: 10 mg/kg CPO
- Group 4: 10 m/kg Sorafenib (positive control)
4.14. Preparation for RNA Sequencing and Analysis
4.15. Molecular Docking Simulation
4.16. Topoisomerase I DNA Relaxation Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruix, J.; Sherman, M. Practice Guidelines Committee, American Association for the Study of Liver Diseases, Management of hepatocellular carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.; Klein, A. AASLD single-topic research conference on hepatocellular carcinoma: Conference proceedings. Hepatology 2004, 40, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Chiaramonte, M.; Stroffolini, T.; Vian, A.; Stazi, M.A.; Floreani, A.; Lorenzoni, U.; Lobello, S.; Farinati, F.; Naccarato, R. Rate of incidence of hepatocellular carcinoma in patients with compensated viral cirrhosis. Cancer 1999, 85, 2132–2137. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.W.; Song, Y.; Kim, S.H.; Kim, J.S.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. CD133 confers cancer stem-like cell properties by stabilizing EGFR-AKT signaling in hepatocellular carcinoma. Cancer Lett. 2017, 389, 1–10. [Google Scholar] [CrossRef]
- Giampieri, R.; Scartozzi, M.; Loretelli, C.; Piva, F.; Mandolesi, A.; Lezoche, G.; Del Prete, M.; Bittoni, A.; Faloppi, L.; Bianconi, M.; et al. Cancer stem cell gene profile as predictor of relapse in high risk stage II and stage III, radically resected colon cancer patients. PLoS ONE 2013, 8, e72843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A.; Kamiyama, T.; Yokoo, H.; Nakanishi, K.; Kubota, K.; Haga, H.; Matsushita, M.; Ozaki, M.; Matsuno, Y.; Todo, S. Cytoplasmic expression of CD133 is an important risk factor for overall survival in hepatocellular carcinoma. Oncol. Rep. 2010, 24, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeng, R.S. Roles of Tumor Microenvironment in Hepatocelluar Carcinoma. Current Cancer Ther. Rev. 2015, 11, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Zhang, M.; Cheng, K.; Li, P.; Han, S.; Li, R.; Su, M.; Zeng, W.; Liu, J.; Guo, J.; et al. Resistance of glioma cells to nutrient-deprived microenvironment can be enhanced by CD133-mediated autophagy. Oncotarget 2016, 7, 76238–76249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.J.; Zhang, S.S.; Guo, X.L.; Sun, K.; Han, Z.P.; Li, R.; Zhao, Q.D.; Deng, W.J.; Xie, X.Q.; Zhang, J.W.; et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013, 339, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Luo, Z.; Dong, L.; Tan, Y.; Yang, J.; Feng, G.; Wu, M.; Li, Z.; Wang, H. CD133/prominin-1-mediated autophagy and glucose uptake beneficial for hepatoma cell survival. PLoS ONE 2013, 8, e56878. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Jang, J.; Shin, T.H.; Bae, S.M.; Kim, J.S.; Kim, K.M.; Myung, S.J.; Choi, E.K.; Seo, H.R. Sulfasalazine attenuates evading anticancer response of CD133-positive hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2017, 36, 38. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.W.; Song, Y.; Kim, S.H.; Kim, J.; Seo, H.R. Potential mechanisms of CD133 in cancer stem cells. Life Sci. 2017, 184, 25–29. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acid. Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Mak, A.B.; Nixon, A.M.; Kittanakom, S.; Stewart, J.M.; Chen, G.I.; Curak, J.; Gingras, A.C.; Mazitschek, R.; Neel, B.G.; Stagljar, I.; et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012, 2, 951–963. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Hayward, R.L.; Meng, L.H.; Takemura, H.; Aune, G.J.; Bonner, W.M.; Aladjem, M.I.; Kohn, K.W.; Pommier, Y. p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene 2006, 25, 2839–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, R.J.; Fiorani, P.; Sugawara, M.; Bjornsti, M.A. CDC45 and DPB11 are required for processive DNA replication and resistance to DNA topoisomerase I-mediated DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 11440–11445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackbarth, J.S.; Galvez-Peralta, M.; Dai, N.T.; Loegering, D.A.; Peterson, K.L.; Meng, X.W.; Karnitz, L.M.; Kaufmann, S.H. Mitotic phosphorylation stimulates DNA relaxation activity of human topoisomerase I. J. Boil. Chem. 2008, 283, 16711–16722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, K.B.; Hadaschik, D.; Virtue, S.; Cawthorn, W.P.; Ridley, S.H.; O’Rahilly, S.; Siddle, K. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ. 2009, 16, 782–789. [Google Scholar] [CrossRef]
- Leblanc, S.E.; Srinivasan, R.; Ferri, C.; Mager, G.M.; Gillian-Daniel, A.L.; Wrabetz, L.; Svaren, J. Regulation of cholesterol/lipid biosynthetic genes by Egr2/Krox20 during peripheral nerve myelination. J. Neurochem. 2005, 93, 737–748. [Google Scholar] [CrossRef]
- Zhang, G.M.; Goyal, H.; Zhang, G.M.; Ma, X.B.; Zhou, Y.T. The “bad” cholesterol can predict abnormal apolipoprotein B levels in a large unselected outpatient cohort. Oncotarget 2018, 9, 8011–8015. [Google Scholar] [CrossRef] [Green Version]
- Farese, R.V., Jr.; Linton, M.F.; Young, S.G. Apolipoprotein B gene mutations affecting cholesterol levels. J. Intern. Med. 1992, 231, 643–652. [Google Scholar] [CrossRef]
- Collins, S. beta-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. 2011, 2, 102. [Google Scholar] [CrossRef] [Green Version]
- Lafontan, M.; Berlan, M. Fat cell adrenergic receptors and the control of white and brown fat cell function. J. Lipid Res. 1993, 34, 1057–1091. [Google Scholar]
- Xie, C.; Powell, C.; Yao, M.; Wu, J.; Dong, Q. Ubiquitin-conjugating enzyme E2C: A potential cancer biomarker. Int. J. Biochem. Cell Boil. 2014, 47, 113–117. [Google Scholar] [CrossRef]
- Ieta, K.; Ojima, E.; Tanaka, F.; Nakamura, Y.; Haraguchi, N.; Mimori, K.; Inoue, H.; Kuwano, H.; Mori, M. Identification of overexpressed genes in hepatocellular carcinoma, with special reference to ubiquitin-conjugating enzyme E2C gene expression. Int. J. Cancer 2007, 121, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kim, I.K.; Choi, I.; Kim, S.H.; Seo, H.R. Oxytetracycline have the therapeutic efficiency in CD133(+) HCC population through suppression CD133 expression by decreasing of protein stability of CD133. Sci. Rep. 2018, 8, 16100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.C.; Hung, M.C. Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors. Clin. Cancer Res. 2009, 15, 6484–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, I.G. Nuclear translocation and functions of growth factor receptors. Semin. Cell Dev. Biol. 2012, 23, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Liao, H.J. Receptor tyrosine kinases in the nucleus. Cold Spring Harb. Perspect. Biol. 2013, 5, a008979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunukova, A.; Neradil, J.; Skoda, J.; Jaros, J.; Hampl, A.; Sterba, J.; Veselska, R. Atypical nuclear localization of CD133 plasma membrane glycoprotein in rhabdomyosarcoma cell lines. Int. J. Mol. Med. 2015, 36, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Wacker, S.A.; Houghtaling, B.R.; Elemento, O.; Kapoor, T.M. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol. 2012, 8, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Schenone, M.; Dancik, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, Z.; Figler, B.; Zhang, H.; Cheng, F. Introduction to RNA-Seq and its applications to drug discovery and development. Drug Dev. Res. 2014, 75, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.H.; Gao, L.; Zhu, X.F.; Wei, K.L.; Zeng, J.J.; Chen, G.; Feng, Z.B. The clinicopathological significance of UBE2C in breast cancer: A study based on immunohistochemistry, microarray and RNA-sequencing data. Cancer Cell Int. 2017, 17, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Ikeda, H.; Taira, N.; Hatoh, S.; Naito, M.; Doihara, H. Overexpression of UbcH10 alternates the cell cycle profile and accelerate the tumor proliferation in colon cancer. BMC Cancer 2009, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Jiang, X.; Zeng, C.; Zheng, S.; Luo, B.; Zeng, Y.; Ding, R.; Jiang, H.; He, Q.; Guo, J.; et al. High expression of ubiquitin-conjugating enzyme 2C (UBE2C) correlates with nasopharyngeal carcinoma progression. BMC Cancer 2013, 13, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, Y.; Ozaki, T.; Miyazaki, K.; Aoyama, M.; Miyazaki, M.; Nakagawara, A. UbcH10 is the cancer-related E2 ubiquitin-conjugating enzyme. Cancer Res. 2003, 63, 4167–4173. [Google Scholar]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acid. Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Bose, M.V.; Gopisetty, G.; Selvaluxmy, G.; Rajkumar, T. Dominant negative Ubiquitin-conjugating enzyme E2C sensitizes cervical cancer cells to radiation. Int. J. Radiat. Boil. 2012, 88, 629–634. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, C.; Rorick, A.; Wu, D.; Chiu, M.; Thomas-Ahner, J.; Chen, Z.; Chen, H.; Clinton, S.K.; Chan, K.K.; et al. CCI-779 inhibits cell-cycle G2-M progression and invasion of castration-resistant prostate cancer via attenuation of UBE2C transcription and mRNA stability. Cancer Res. 2011, 71, 4866–4876. [Google Scholar] [CrossRef] [Green Version]
- Sabitha, K.; Rajkumar, T. Identification of small molecule inhibitors against UBE2C by using docking studies. Bioinformation 2012, 8, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Aradillas-Garc, C.; Cruz, M.; Perez-Luque, E.; Garay-Sevilla, M.E.; Malacara, J.M.; R, A.; Peralta, J.; Burguete-Garcia, A.; Alegria-Torres, J.A. Obesity is associated with the Arg389Gly ADRB1 but not with the Trp64Arg ADRB3 polymorphism in children from San Luis PotosI and Leon, Mexico. J. Biomed. Res. 2016, 31, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Tikhonoff, V.; Hasenkamp, S.; Kuznetsova, T.; Thijs, L.; Jin, Y.; Richart, T.; Zhang, H.; Brand-Herrmann, S.M.; Brand, E.; Casiglia, E.; et al. Blood pressure and metabolic phenotypes in relation to the ADRB1 Arg389Gly and ADRA2B I/D polymorphisms in a White population. J. Hum. Hypertens. 2008, 22, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Velasco, B.I.; Cruz, M.; Madrid-Marina, V.; Martinez-Nava, G.A.; Gomez-Zamudio, J.; Burguete-Garcia, A.I. IRS1, TCF7L2, ADRB1, PPARG, and HHEX polymorphisms associated with atherogenic risk in Mexican population. BioMed Res. Int. 2013, 2013, 394523. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Full Name | Position | Ref. | Alt. | Quality * | Mutation |
|---|---|---|---|---|---|---|
| ADRB1 | Adrenoceptor beta 1 | 114045297 | G | C | 149.9 | G389R |
| APOB | Apolipoprotein B | 21008652 | G | A | 9889.7 | P2739L |
| EGR2 | Early growth response 2 | 62813452 | T | C | 31.77 | K396E |
| 62813453 | C | T | 31.77 | K396E | ||
| UBE2C | Ubiquitin-conjugating enzyme E2C | 45812764 | C | G | 813.77 | S23R |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Kim, S.; Lee, H.; No, J.H.; Ryu, H.C.; Kim, J.; Lim, J.W.; Kim, M.; Choi, I.; Seo, H.R. Chromenopyrimidinone Controls Stemness and Malignancy by suppressing CD133 Expression in Hepatocellular Carcinoma. Cancers 2020, 12, 1193. https://doi.org/10.3390/cancers12051193
Song Y, Kim S, Lee H, No JH, Ryu HC, Kim J, Lim JW, Kim M, Choi I, Seo HR. Chromenopyrimidinone Controls Stemness and Malignancy by suppressing CD133 Expression in Hepatocellular Carcinoma. Cancers. 2020; 12(5):1193. https://doi.org/10.3390/cancers12051193
Chicago/Turabian StyleSong, Yeonhwa, Sanghwa Kim, Hyeryon Lee, Joo Hwan No, Hyung Chul Ryu, Jason Kim, Jee Woong Lim, Moonhee Kim, Inhee Choi, and Haeng Ran Seo. 2020. "Chromenopyrimidinone Controls Stemness and Malignancy by suppressing CD133 Expression in Hepatocellular Carcinoma" Cancers 12, no. 5: 1193. https://doi.org/10.3390/cancers12051193