Mesenchymal Stem Cells As Guideposts for Nanoparticle-Mediated Targeted Drug Delivery in Ovarian Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Physical Characteristics of Nanoparticles
2.2. Kinetics of In Vitro Drug Release
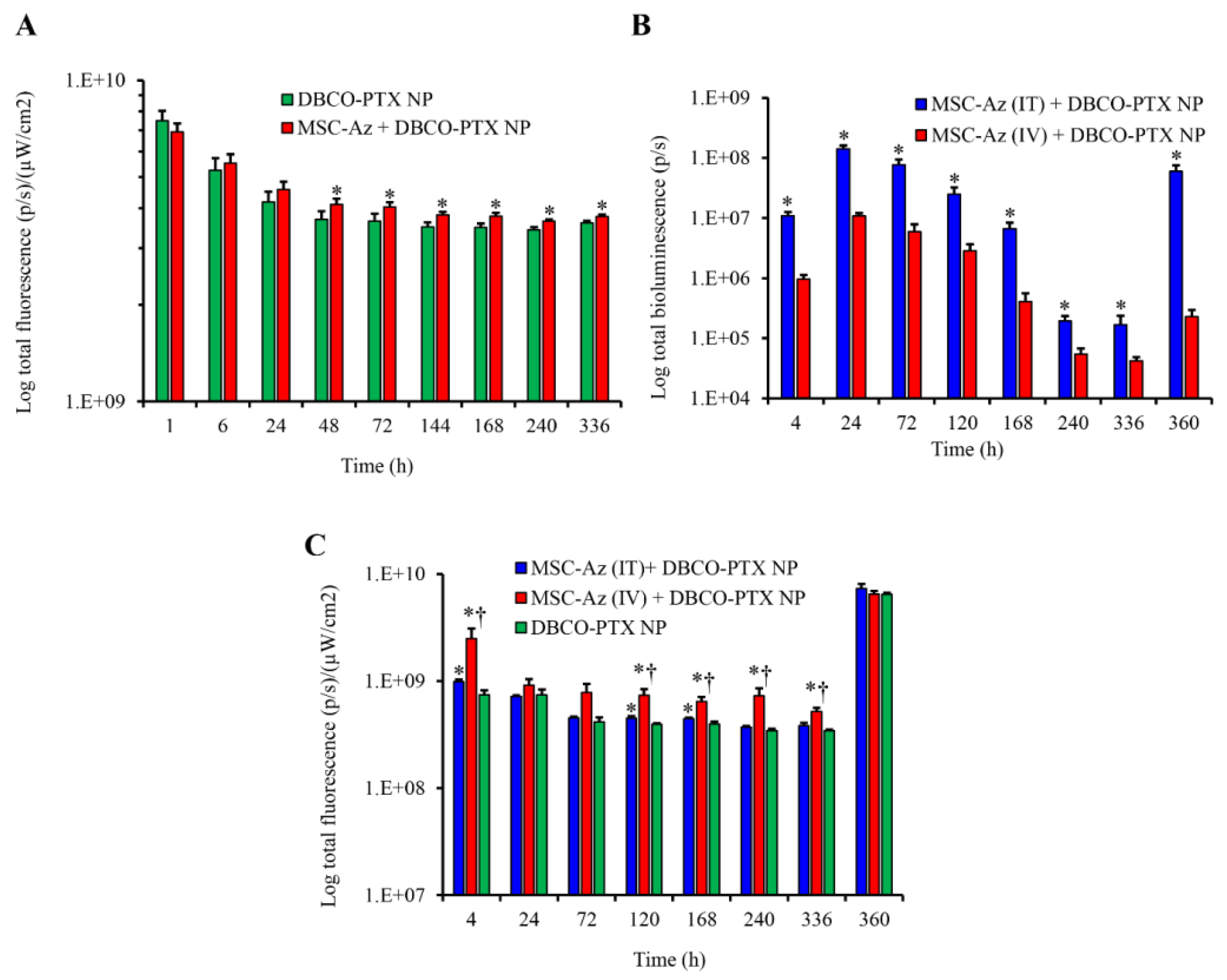
2.3. Biodistribution of Nanoparticles
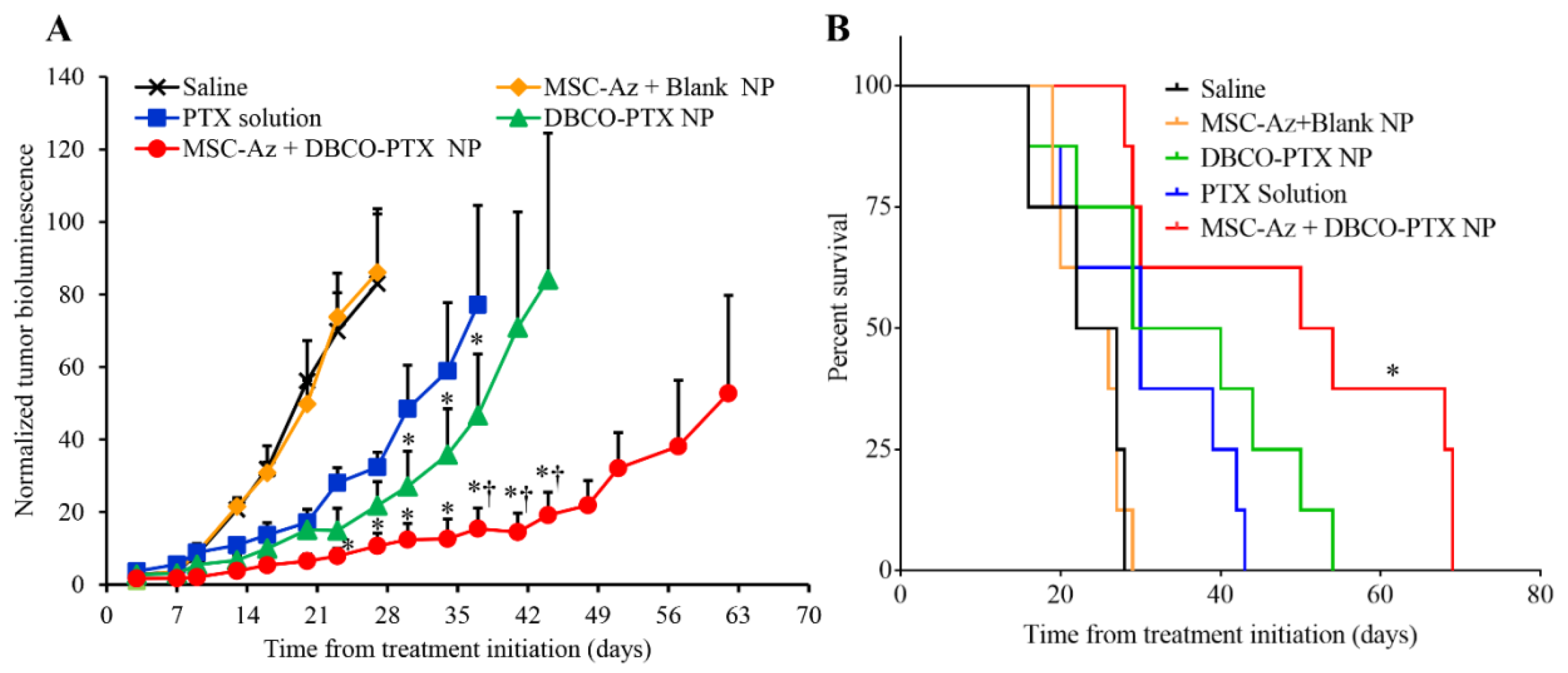
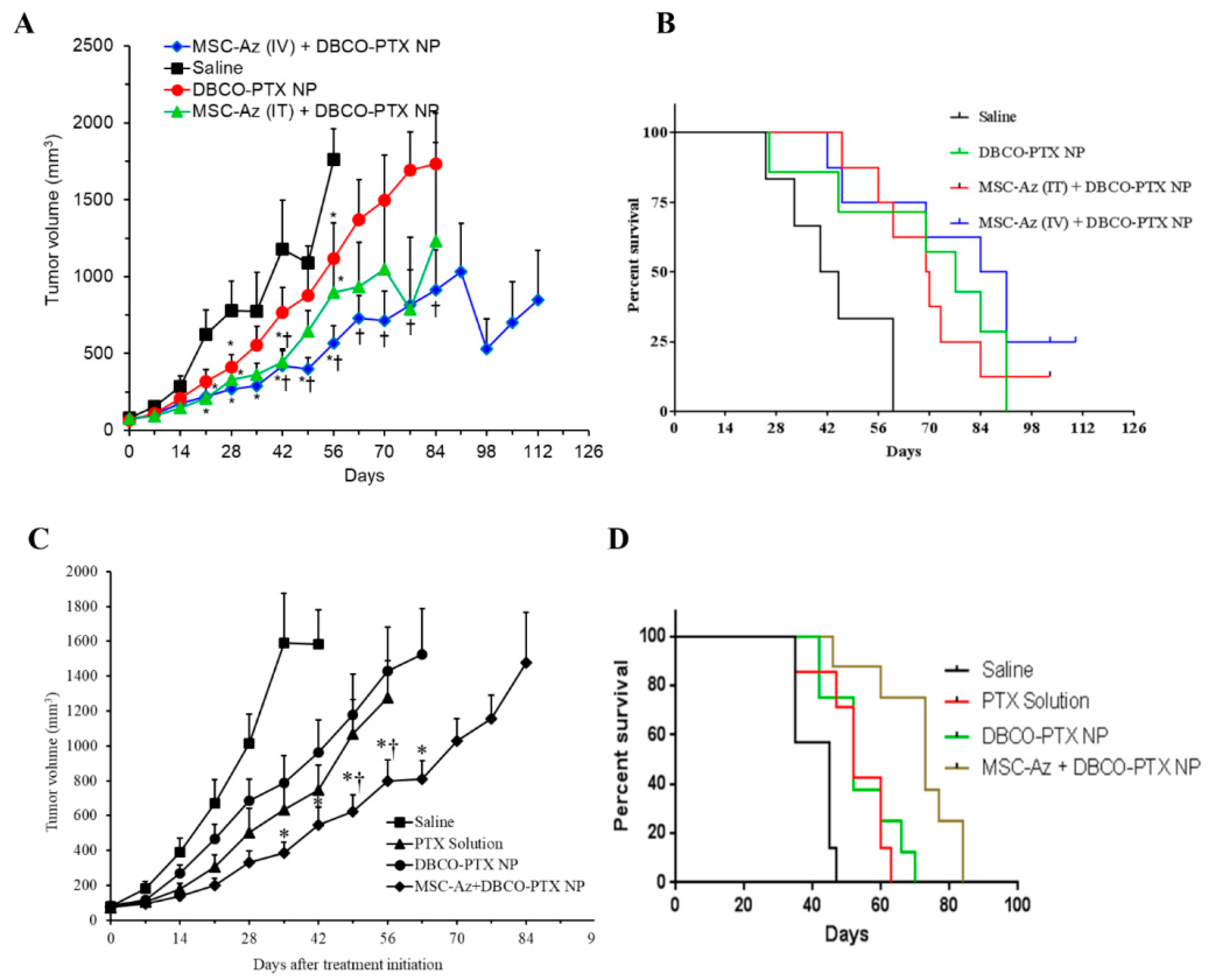
2.4. Anticancer Efficacy of Two-Step Targeting Approach
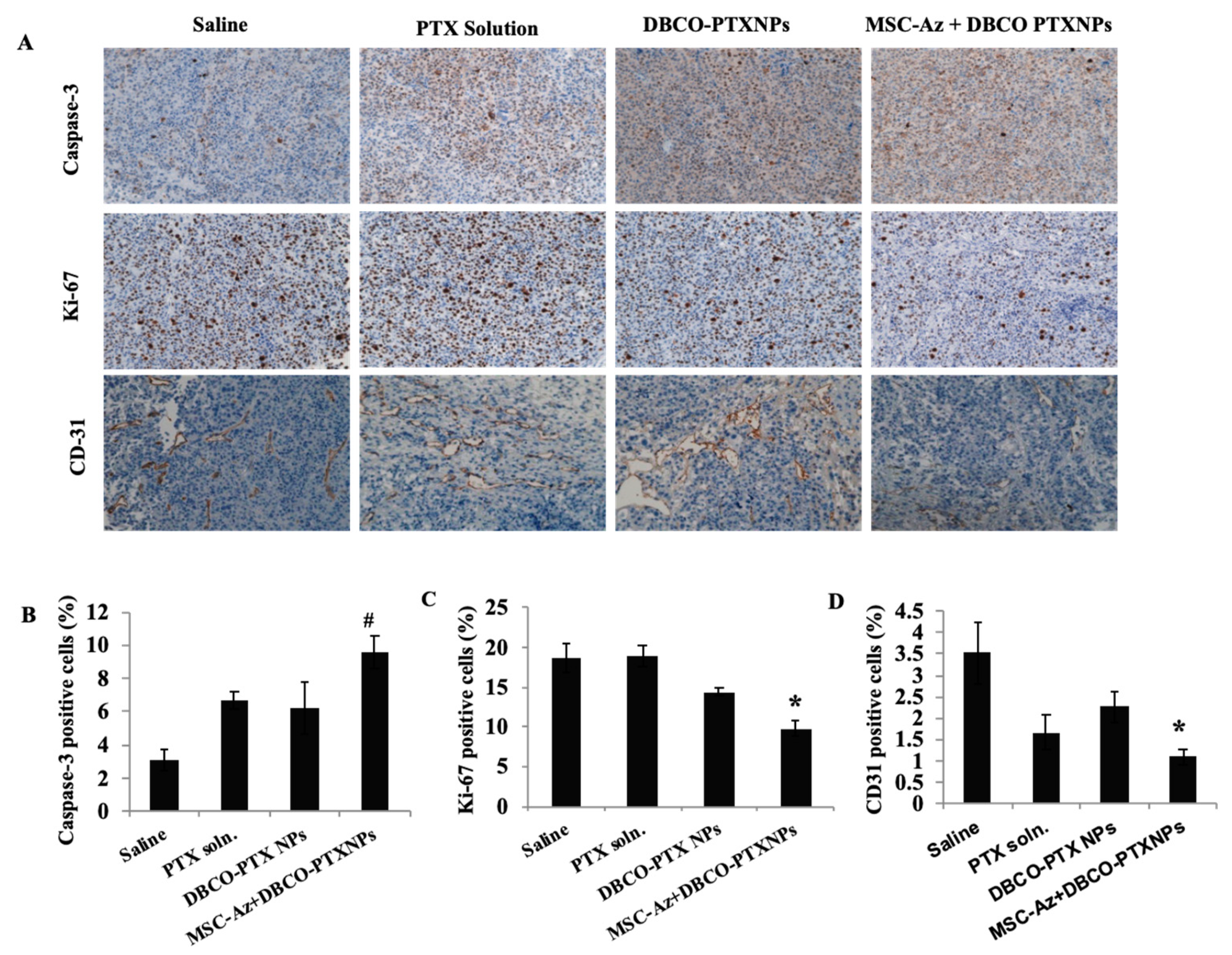
2.5. Immunohistochemical Analysis of Tumors
2.6. Real-Time PCR
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of PLA-PEG-DBCO
4.3. Preparation of Nanoparticles
4.4. Characterization of Nanoparticles
4.5. In Vitro Drug Release
4.6. Cell Culture
4.7. Generation of Luciferase-Expressing C200 (C200-Luc) Cells and MSCs (MSC-Luc)
4.8. Generation of Azide Labeled MSCs (MSC-Az)
4.9. Animal Tumor Models
4.9.1. Generation of Orthotopic Ovarian Tumor Model
4.9.2. Generation of PDX Tumor Models
4.9.3. Biodistribution and Efficacy Studies were Performed in the PDX Ovarian Tumor Models as Described Below
4.10. Biodistribution of Nanoparticles
4.10.1. Orthotopic Ovarian Tumor Model
4.10.2. PDX Tumor Models
4.11. Anticancer Efficacy of Two-Step Targeting Approach
4.11.1. Efficacy against C200-Luc Tumors
4.11.2. Efficacy against PDX Tumors
4.11.3. Effect of Increased Dose of MSC against PDX Tumors
4.12. Immunohistochemical Analysis of Tumors
4.13. Real Time PCR
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stewart, S.L.; Harewood, R.; Matz, M.; Rim, S.H.; Sabatino, S.A.; Ward, K.C.; Weir, H.K. Disparities in ovarian cancer survival in the United States (2001–2009): Findings from the CONCORD-2 study. Cancer 2017, 123 (Suppl. S24), 5138–5159. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markman, M. Current standards of care for chemotherapy of optimally cytoreduced advanced epithelial ovarian cancer. Gynecol. Oncol. 2013, 131, 241–245. [Google Scholar] [CrossRef]
- Vergote, I.; Trope, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N.; Verheijen, R.H.; van der Burg, M.E.; Lacave, A.J.; Panici, P.B.; et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ren, W.; Zhong, T.; Zhang, S.; Huang, D.; Guo, Y.; Yao, X.; Wang, C.; Zhang, W.Q.; Zhang, X.; et al. Tumor-specific pH-responsive peptide-modified pH-sensitive liposomes containing doxorubicin for enhancing glioma targeting and anti-tumor activity. J. Control. Release 2016, 222, 56–66. [Google Scholar] [CrossRef]
- Kieler-Ferguson, H.M.; Chan, D.; Sockolosky, J.; Finney, L.; Maxey, E.; Vogt, S.; Szoka, F.C., Jr. Encapsulation, controlled release, and antitumor efficacy of cisplatin delivered in liposomes composed of sterol-modified phospholipids. Eur. J. Pharm. Sci. 2017, 103, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.W.; Chun, M.K.; Kim, O.; Subedi, R.K.; Ahn, S.G.; Yoon, J.H.; Choi, H.K. Doxorubicin-loaded solid lipid nanoparticles to overcome multidrug resistance in cancer therapy. Nanomedicine 2010, 6, 210–213. [Google Scholar] [CrossRef]
- Hu, J.; Fu, S.; Peng, Q.; Han, Y.; Xie, J.; Zan, N.; Chen, Y.; Fan, J. Paclitaxel-loaded polymeric nanoparticles combined with chronomodulated chemotherapy on lung cancer: In vitro and in vivo evaluation. Int. J. Pharm. 2017, 516, 313–322. [Google Scholar] [CrossRef]
- Yellepeddi, V.K.; Kumar, A.; Maher, D.M.; Chauhan, S.C.; Vangara, K.K.; Palakurthi, S. Biotinylated PAMAM dendrimers for intracellular delivery of cisplatin to ovarian cancer: Role of SMVT. Anticancer Res. 2011, 31, 897–906. [Google Scholar]
- Zhong, Q.; Bielski, E.R.; Rodrigues, L.S.; Brown, M.R.; Reineke, J.J.; da Rocha, S.R. Conjugation to poly(amidoamine) dendrimers and pulmonary delivery reduce cardiac accumulation and enhance antitumor activity of doxorubicin in lung metastasis. Mol. Pharm. 2016, 13, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- Son, G.M.; Kim, H.Y.; Ryu, J.H.; Chu, C.W.; Kang, D.H.; Park, S.B.; Jeong, Y.I. Self-assembled polymeric micelles based on hyaluronic acid-g-poly(D,L-lactide-co-glycolide) copolymer for tumor targeting. Int. J. Mol. Sci. 2014, 15, 16057–16068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzano, G.; Navarro, G.; Trivedi, M.S.; De Rosa, G.; Torchilin, V.P. Multifunctional polymeric micelles co-loaded with anti-survivin siRNA and paclitaxel overcome drug resistance in an animal model of ovarian cancer. Mol. Cancer 2015, 14, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Florence, A.T. “Targeting” nanoparticles: The constraints of physical laws and physical barriers. J. Control. Release 2012, 164, 115–124. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Lazarovits, J.; Chen, Y.Y.; Sykes, E.A.; Chan, W.C. Nanoparticle-blood interactions: The implications on solid tumour targeting. Chem. Commun. (Camb.) 2015, 51, 2756–2767. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. Odyssey of a cancer nanoparticle: From injection site to site of action. Nano Today 2012, 7, 606–618. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Termsarasab, U.; Park, J.H.; Lee, S.Y.; Ko, S.H.; Shim, J.S.; Chung, S.J.; Cho, H.J.; Kim, D.D. Dual CD44 and folate receptor-targeted nanoparticles for cancer diagnosis and anticancer drug delivery. J. Control. Release 2016, 236, 38–46. [Google Scholar] [CrossRef]
- Song, X.; Ren, Y.; Zhang, J.; Wang, G.; Han, X.; Zheng, W.; Zhen, L. Targeted delivery of doxorubicin to breast cancer cells by aptamer functionalized DOTAP/DOPE liposomes. Oncol. Rep. 2015, 34, 1953–1960. [Google Scholar] [CrossRef] [Green Version]
- Valetti, S.; Maione, F.; Mura, S.; Stella, B.; Desmaele, D.; Noiray, M.; Vergnaud, J.; Vauthier, C.; Cattel, L.; Giraudo, E.; et al. Peptide-functionalized nanoparticles for selective targeting of pancreatic tumor. J. Control. Release 2014, 192, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Sadhukha, T.; Prabha, S. Glycoengineered mesenchymal stem cells as an enabling platform for two-step targeting of solid tumors. Biomaterials 2016, 88, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-W.; Lee, S.; Na, J.H.; Yoon, H.I.; Lee, D.-E.; Koo, H.; Cho, Y.W.; Kim, S.H.; Jeong, S.Y.; Kwon, I.C.; et al. Cell labeling and tracking method without distorted signals by phagocytosis of macrophages. Theranostics 2014, 4, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.; Lee, S.; Na, J.H.; Kim, S.H.; Hahn, S.K.; Choi, K.; Kwon, I.C.; Jeong, S.Y.; Kim, K. Bioorthogonal copper-free click chemistry in vivo for tumor-targeted delivery of nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 11836–11840. [Google Scholar] [CrossRef]
- Best, M.D. Click chemistry and bioorthogonal reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Muggia, F.M.; Braly, P.S.; Brady, M.F.; Sutton, G.; Niemann, T.H.; Lentz, S.L.; Alvarez, R.D.; Kucera, P.R.; Small, J.M. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2000, 18, 106–115. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Vasanthakumar, S.; Bende, G.; Snehalatha, M. Nanoparticulate drug delivery systems for cancer chemotherapy. Mol. Membr. Biol. 2010, 27, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Javan, M.R.; Khosrojerdi, A.; Moazzeni, S.M. New insights into implementation of mesenchymal stem cells in cancer therapy: Prospects for anti-angiogenesis treatment. Front. Oncol. 2019, 9, 840. [Google Scholar] [CrossRef]
- Ning, X.; Guo, J.; Wolfert, M.A.; Boons, G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. 2008, 47, 2253–2255. [Google Scholar] [CrossRef]
- Lee, S.; Koo, H.; Na, J.H.; Han, S.J.; Min, H.S.; Lee, S.J.; Kim, S.H.; Yun, S.H.; Jeong, S.Y.; Kwon, I.C.; et al. Chemical tumor-targeting of nanoparticles based on metabolic glycoengineering and click chemistry. ACS Nano 2014, 8, 2048–2063. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Anker, P.S.I.; Scherjon, S.A.; Kleijburg-van der Keur, C.; de Groot-Swings, G.M.J.S.; Claas, F.H.J.; Fibbe, W.E.; Kanhai, H.H.H. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells 2004, 22, 1338–1345. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Koneru, R.; Glod, J.; Banerjee, D. The JAK2/STAT3- mediated SDF-1/CXCR4 axis promotes mesenchymal stem cell migration in response to the tumor microenvironment. Cancer Res. 2007, 67, 573. [Google Scholar]
- Kalimuthu, S.; Zhu, L.; Oh, J.M.; Gangadaran, P.; Lee, H.W.; Baek, S.H.; Rajendran, R.L.; Gopal, A.; Jeong, S.Y.; Lee, S.-W.; et al. Migration of mesenchymal stem cells to tumor xenograft models and in vitro drug delivery by doxorubicin. Int. J. Med. Sci. 2018, 15, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Meledeo, M.A.; Wang, Z.; Khanna, H.S.; Paruchuri, V.D.P.; Yarema, K.J. Metabolic glycoengineering: Sialic acid and beyond. Glycobiology 2009, 19, 1382–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layek, B.; Sehgal, D.; Argenta, P.A.; Panyam, J.; Prabha, S. Nanoengineering of mesenchymal stem cells via surface modification for efficient cancer therapy. Adv. Ther. 2019, 2, 1900043. [Google Scholar] [CrossRef]
- Moku, G.; Layek, B.; Trautman, L.; Putnam, S.; Panyam, J.; Prabha, S. Improving payload capacity and anti-tumor efficacy of mesenchymal stem cells using TAT peptide functionalized polymeric nanoparticles. Cancers 2019, 11, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, O.J.; Holland, E.C. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006, 66, 3355–3359. [Google Scholar] [CrossRef] [Green Version]
- Ricci, F.; Broggini, M.; Damia, G. Revisiting ovarian cancer preclinical models: Implications for a better management of the disease. Cancer Treat. Rev. 2013, 39, 561–568. [Google Scholar] [CrossRef]
- Sausville, E.A.; Burger, A.M. Contributions of human tumor xenografts to anticancer drug development. Cancer Res. 2006, 66, 3351–3354. [Google Scholar] [CrossRef] [Green Version]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J., Jr.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef] [Green Version]
- Elkas, J.C.; Baldwin, R.L.; Pegram, M.; Tseng, Y.; Slamon, D.; Karlan, B.Y. A human ovarian carcinoma murine xenograft model useful for preclinical trials. Gynecol. Oncol. 2002, 87, 200–206. [Google Scholar] [CrossRef]
- Press, J.Z.; Kenyon, J.A.; Xue, H.; Miller, M.A.; De Luca, A.; Miller, D.M.; Huntsman, D.G.; Gilks, C.B.; McAlpine, J.N.; Wang, Y.Z. Xenografts of primary human gynecological tumors grown under the renal capsule of NOD/SCID mice show genetic stability during serial transplantation and respond to cytotoxic chemotherapy. Gynecol. Oncol. 2008, 110, 256–264. [Google Scholar] [CrossRef]
- Weroha, S.J.; Becker, M.A.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; AlHilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG oncology/gynecologic oncology group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Eva, A.; Robbins, K.C.; Andersen, P.R.; Srinivasan, A.; Tronick, S.R.; Reddy, E.P.; Ellmore, N.W.; Galen, A.T.; Lautenberger, J.A.; Papas, T.S.; et al. Cellular genes analogous to retroviral onc genes are transcribed in human tumour cells. Nature 1982, 295, 116–119. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [Green Version]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef] [Green Version]
- Patil, Y.B.; Toti, U.S.; Khdair, A.; Ma, L.; Panyam, J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials 2009, 30, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadhukha, T.; O’Brien, T.D.; Prabha, S. Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J. Control. Release 2014, 196, 243–251. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Values |
|---|---|
| Particle size (nm) | 331 ± 25.1 |
| PDI | 0.22 ± 0.03 |
| Zeta potential (mV) | −11.5 ± 1.3 |
| Drug loading (%) | 17.2 ± 0.8 |
| Entrapment efficiency (%) | 73.8 ± 3.6 |
| Group/Score | Ki-67 | Caspase-3 | CD31 | Necrosis Score | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Saline | 0 | 0 | 0 | 3 | 3 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| DBCO-PTX NP | 0 | 1 | 1 | 2 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 1 | 1 | 2 |
| MSC-Az (IV) + DBCO-PTX NP | 0 | 0 | 2 | 2 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 1 | 3 |
| MSC-Az (IT) + DBCO-PTX NP | 0 | 1 | 1 | 2 | 4 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 1 | 0 | 3 |
| Sample | Expression Fold Change (2−∆∆Ct) |
|---|---|
| MSC-Az (IV) + DBCO-PTX NP | 1.90 ± 0.92 |
| MSC-Az (IT) + DBCO-PTX NP | 1.81 ± 0.56 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Layek, B.; Shetty, M.; Nethi, S.K.; Sehgal, D.; Starr, T.K.; Prabha, S. Mesenchymal Stem Cells As Guideposts for Nanoparticle-Mediated Targeted Drug Delivery in Ovarian Cancer. Cancers 2020, 12, 965. https://doi.org/10.3390/cancers12040965
Layek B, Shetty M, Nethi SK, Sehgal D, Starr TK, Prabha S. Mesenchymal Stem Cells As Guideposts for Nanoparticle-Mediated Targeted Drug Delivery in Ovarian Cancer. Cancers. 2020; 12(4):965. https://doi.org/10.3390/cancers12040965
Chicago/Turabian StyleLayek, Buddhadev, Mihir Shetty, Susheel Kumar Nethi, Drishti Sehgal, Timothy K. Starr, and Swayam Prabha. 2020. "Mesenchymal Stem Cells As Guideposts for Nanoparticle-Mediated Targeted Drug Delivery in Ovarian Cancer" Cancers 12, no. 4: 965. https://doi.org/10.3390/cancers12040965