Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments

 ,
,  ,
,
Abstract
:1. Introduction
2. Pathogenesis
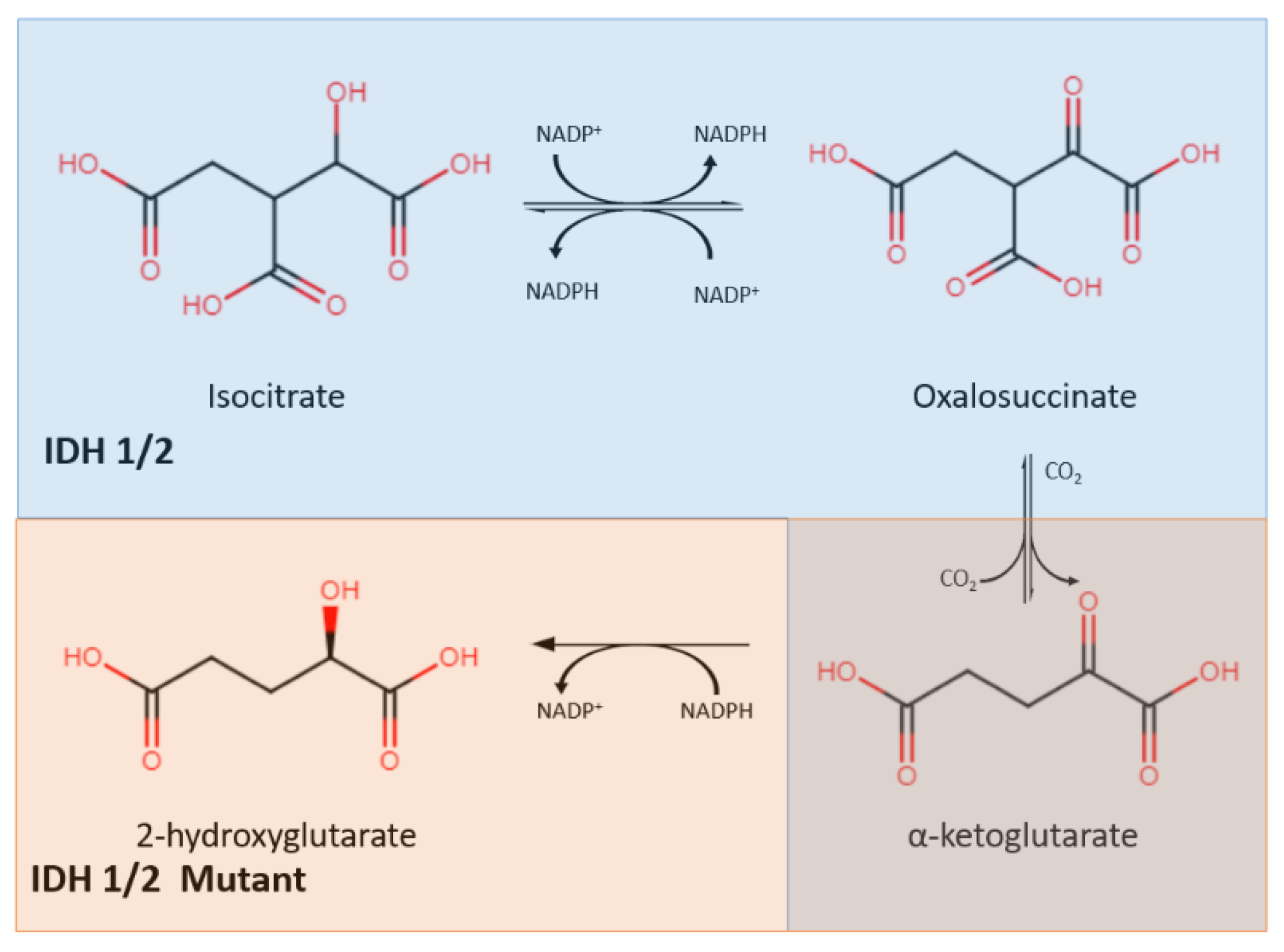
2.1. IDH Mutation
2.2. Notch Pathway
2.3. Ceramide Signaling
2.4. Vascular Endothelial Growth Factor (VEGF) Signaling Pathway
2.5. PDGF Signaling
2.6. Epidermal Growth Factor Receptor (EGFR) Pathway
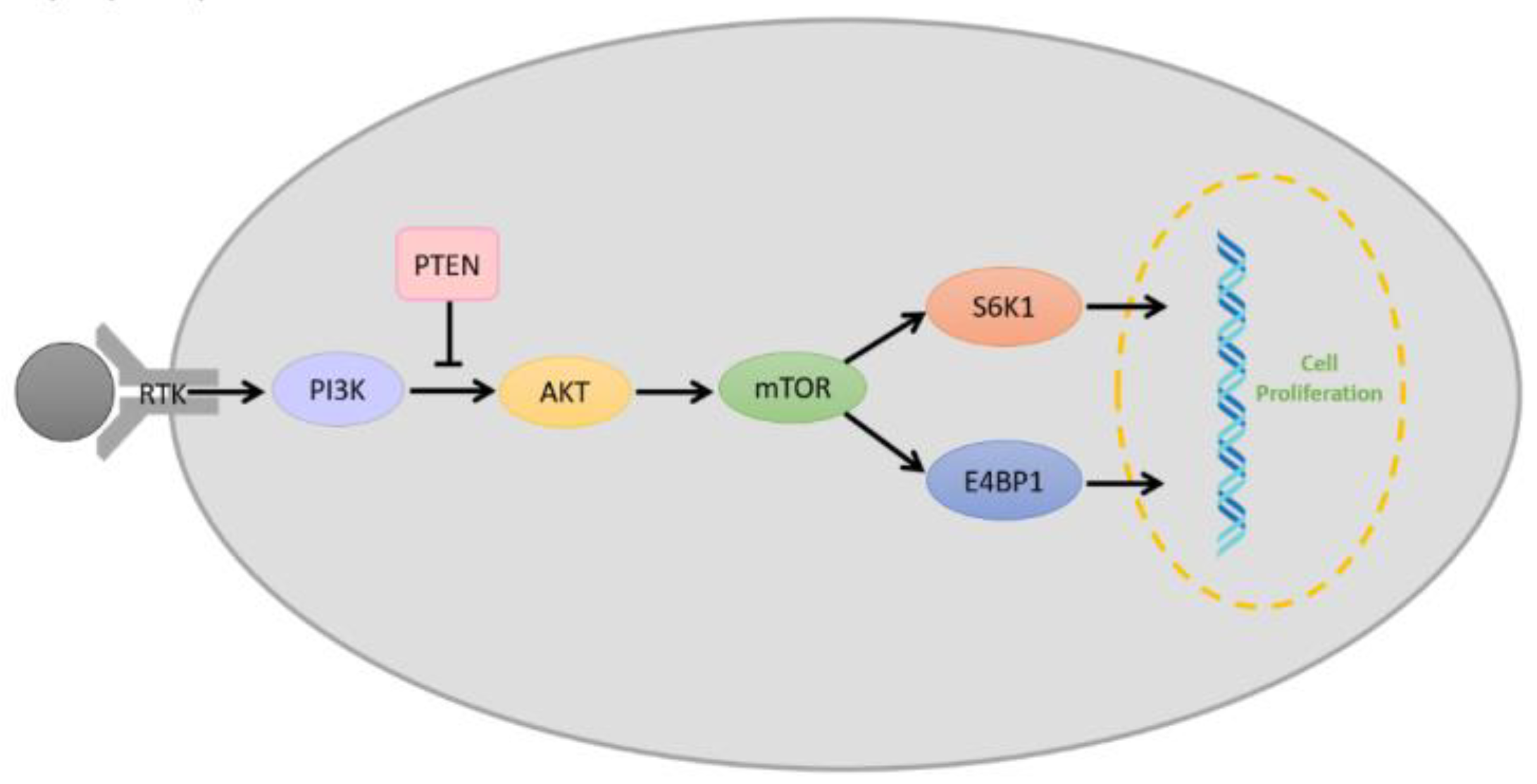
2.7. PI3K/AKT/mTOR Pathway
2.8. Phosphate and Tensin Homolog (PTEN) Signaling
2.9. SHH Signaling
3. Current Chemotherapeutic Development
3.1. FDA-Approved Chemotherapeutic Agents
3.2. Published Clinical Trials
3.3. Ongoing Clinical Trials
4. Novel Therapies
4.1. Laser Interstitial Thermal Therapy (LITT)
4.2. Tumor Treating Fields (TTFields)
4.3. Immunotherapy
4.3.1. Immune Checkpoint Inhibitors
4.3.2. T-Cell Therapy
4.3.3. Viral Therapy
4.3.4. Vaccine Therapy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, K.; Shagufta, B.I.; Abbas, S.Q.; Patel, S.; Khan, I.; Shah, S.A.A.; Akhter, N.; Hassan, S.S.U. Current status and future therapeutic perspectives of glioblastoma multiforme (GBM) therapy: A review. Biomed. Pharmacother. 2017, 92, 681–689. [Google Scholar] [CrossRef]
- Ferguson, S.; Lesniak, M.S. Percival Bailey and the classification of brain tumors. Neurosurg. Focus 2005, 18, e7. [Google Scholar] [CrossRef]
- Zulch, K.J.; Wechsler, W. Pathology and Classification of Gliomas. In Progress in Neurological Surgery; Karger Publisher: Basel, Switzerland, 1968; Volume 2, pp. 1–84. [Google Scholar]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Campian, J.L.; Gujar, A.D.; Tsien, C.; Ansstas, G.; Tran, D.D.; DeWees, T.A.; Lockhart, A.C.; Kim, A.H. Final results of a phase I dose-escalation, dose-expansion study of adding disulfiram with or without copper to adjuvant temozolomide for newly diagnosed glioblastoma. J. Neurooncol. 2018, 138, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Cancer, I.A.f.R.o. WHO Classification of Tumours of the Central Nervous System; WTO: Geneva, Switzerland, 2016; Volume 1. [Google Scholar]
- Stöppler, M.C.; Shiel, W.C.; Credo Reference (Firm); WebMD (Firm). Webster’s New World Medical Dictionary, 3rd ed.; redo Reference: Boston, MA, USA; Wiley: Hoboken, NJ, USA, 2014; p. 1. [Google Scholar]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Kobayashi, H.; Terasaka, S.; Ishii, N.; Ikeda, J.; Kanno, H.; Nishihara, H.; Tanaka, S.; Houkin, K. The impact of extent of resection and histological subtype on the outcome of adult patients with high-grade gliomas. Jpn. J. Clin. Oncol. 2012, 42, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro Oncol. 2002, 4, 278–299. [Google Scholar] [CrossRef]
- Preusser, M.; de Ribaupierre, S.; Wohrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21. [Google Scholar] [CrossRef]
- Aoki, T.; Hashimoto, N.; Matsutani, M. Management of glioblastoma. Expert Opin. Pharmacother. 2007, 8, 3133–3146. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Berger, M.S. Recent surgical management of gliomas. Adv. Exp. Med. Biol. 2012, 746, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Jamshidi, A.; Davis, G.; Sherman, J.H. Current trends in the surgical management and treatment of adult glioblastoma. Ann. Transl. Med. 2015, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Ryken, T.C.; Frankel, B.; Julien, T.; Olson, J.J. Surgical management of newly diagnosed glioblastoma in adults: Role of cytoreductive surgery. J. Neurooncol. 2008, 89, 271–286. [Google Scholar] [CrossRef]
- Barbagallo, G.M.; Jenkinson, M.D.; Brodbelt, A.R. ’Recurrent’ glioblastoma multiforme, when should we reoperate? Br. J. Neurosurg. 2008, 22, 452–455. [Google Scholar] [CrossRef]
- Cabrera, A.R.; Kirkpatrick, J.P.; Fiveash, J.B.; Shih, H.A.; Koay, E.J.; Lutz, S.; Petit, J.; Chao, S.T.; Brown, P.D.; Vogelbaum, M.; et al. Radiation therapy for glioblastoma: Executive summary of an American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. Pract. Radiat. Oncol. 2016, 6, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Minniti, G.; Filippi, A.R.; Osti, M.F.; Ricardi, U. Radiation therapy for older patients with brain tumors. Radiat. Oncol. 2017, 12, 101. [Google Scholar] [CrossRef]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in Radiotherapy for Glioblastoma. Front. Neurol. 2017, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Corso, C.D.; Bindra, R.S.; Mehta, M.P. The role of radiation in treating glioblastoma: Here to stay. J. Neurooncol. 2017, 134, 479–485. [Google Scholar] [CrossRef]
- Fedoy, A.E.; Yang, N.; Martinez, A.; Leiros, H.K.; Steen, I.H. Structural and functional properties of isocitrate dehydrogenase from the psychrophilic bacterium Desulfotalea psychrophila reveal a cold-active enzyme with an unusual high thermal stability. J. Mol. Biol. 2007, 372, 130–149. [Google Scholar] [CrossRef]
- Kaminska, B.; Czapski, B.; Guzik, R.; Krol, S.K.; Gielniewski, B. Consequences of IDH1/2 Mutations in Gliomas and an Assessment of Inhibitors Targeting Mutated IDH Proteins. Molecules 2019, 24, 968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Lino, M.M.; Merlo, A.; Boulay, J.L. Notch signaling in glioblastoma: A developmental drug target? BMC Med. 2010, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Hao, C.; Xiao-Feng, L.; Yu-Chen, L.; Yu-Bin, F.; Lei, Z. Molecular mechanism of Notch signaling with special emphasis on microRNAs: Implications for glioma. J. Cell. Physiol. 2018, 234, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers (Basel) 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, M.; Wang, S.; Sang, Y.; Sun, P.; Lal, B.; Goodwin, C.R.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Laterra, J.; Xia, S. Regulation of glioblastoma stem cells by retinoic acid: Role for Notch pathway inhibition. Oncogene 2011, 30, 3454–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.S.; Awad, A.J.; Shabani, S.; Doan, N. Molecular Targeting of Acid Ceramidase in Glioblastoma: A Review of Its Role, Potential Treatment, and Challenges. Pharmaceutics 2018, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.B.; Nguyen, H.S.; Montoure, A.; Al-Gizawiy, M.M.; Mueller, W.M.; Kurpad, S.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; et al. Acid ceramidase is a novel drug target for pediatric brain tumors. Oncotarget 2017, 8, 24753–24761. [Google Scholar] [CrossRef]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef]
- Wick, W.; Weller, M.; Weiler, M.; Batchelor, T.; Yung, A.W.; Platten, M. Pathway inhibition: Emerging molecular targets for treating glioblastoma. Neuro Oncol. 2011, 13, 566–579. [Google Scholar] [CrossRef]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Tasaki, T.; Nakata, S.; Yamashita, K.; Yoshioka, H.; Izumoto, S.; Kato, A.; Fujita, M. Efficacy of Combination Therapy with MET and VEGF Inhibitors for MET-overexpressing Glioblastoma. Anticancer Res. 2017, 37, 3871–3876. [Google Scholar] [CrossRef] [Green Version]
- Weathers, S.P.; de Groot, J. VEGF Manipulation in Glioblastoma. Oncology (Williston Park) 2015, 29, 720–727. [Google Scholar] [PubMed]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef] [PubMed]
- Mischel, P.S.; Cloughesy, T.F. Targeted molecular therapy of GBM. Brain Pathol. 2003, 13, 52–61. [Google Scholar] [CrossRef]
- Shih, A.H.; Holland, E.C. Platelet-derived growth factor (PDGF) and glial tumorigenesis. Cancer Lett. 2006, 232, 139–147. [Google Scholar] [CrossRef]
- Heldin, C.H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Cantanhede, I.G.; de Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef] [Green Version]
- Westermark, B. Platelet-derived growth factor in glioblastoma-driver or biomarker? Ups. J. Med. Sci. 2014, 119, 298–305. [Google Scholar] [CrossRef]
- Popescu, A.M.; Alexandru, O.; Brindusa, C.; Purcaru, S.O.; Tache, D.E.; Tataranu, L.G.; Taisescu, C.; Dricu, A. Targeting the VEGF and PDGF signaling pathway in glioblastoma treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7825–7837. [Google Scholar]
- Hong, J.D.; Wang, X.; Peng, Y.P.; Peng, J.H.; Wang, J.; Dong, Y.P.; He, D.; Peng, Z.Z.; Tu, Q.S.; Sheng, L.F.; et al. Silencing platelet-derived growth factor receptor-beta enhances the radiosensitivity of C6 glioma cells in vitro and in vivo. Oncol. Lett. 2017, 14, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Cenciarelli, C.; Marei, H.E.; Zonfrillo, M.; Pierimarchi, P.; Paldino, E.; Casalbore, P.; Felsani, A.; Vescovi, A.L.; Maira, G.; Mangiola, A. PDGF receptor alpha inhibition induces apoptosis in glioblastoma cancer stem cells refractory to anti-Notch and anti-EGFR treatment. Mol. Cancer 2014, 13, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Tachibana, O.; Sata, K.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996, 6, 217–223, discussion 223–214. [Google Scholar] [CrossRef]
- Kraus, J.A.; Felsberg, J.; Tonn, J.C.; Reifenberger, G.; Pietsch, T. Molecular genetic analysis of the TP53, PTEN, CDKN2A, EGFR, CDK4 and MDM2 tumour-associated genes in supratentorial primitive neuroectodermal tumours and glioblastomas of childhood. Neuropathol. Appl. Neurobiol. 2002, 28, 325–333. [Google Scholar] [CrossRef]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef] [Green Version]
- Halatsch, M.E.; Gehrke, E.E.; Vougioukas, V.I.; Botefur, I.C.; A-Borhani, F.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef]
- Reardon, D.A.; Groves, M.D.; Wen, P.Y.; Nabors, L.; Mikkelsen, T.; Rosenfeld, S.; Raizer, J.; Barriuso, J.; McLendon, R.E.; Suttle, A.B.; et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin. Cancer Res. 2013, 19, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 9230479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballo, G.B.; Honorato, J.R.; de Lopes, G.P.F.; Spohr, T. A highlight on Sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Mantamadiotis, T. Towards Targeting PI3K-Dependent Regulation of Gene Expression in Brain Cancer. Cancers (Basel) 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Lino, M.M.; Merlo, A. PI3Kinase signaling in glioblastoma. J. Neurooncol. 2011, 103, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Janbazian, L.; Karamchandani, J.; Das, S. Mouse models of glioblastoma: Lessons learned and questions to be answered. J. Neurooncol. 2014, 118, 1–8. [Google Scholar] [CrossRef]
- Romano, C.; Schepis, C. PTEN gene: A model for genetic diseases in dermatology. ScientificWorldJournal 2012, 2012, 252457. [Google Scholar] [CrossRef] [Green Version]
- Lester, A.; Rapkins, R.; Nixdorf, S.; Khasraw, M.; McDonald, K. Combining PARP inhibitors with radiation therapy for the treatment of glioblastoma: Is PTEN predictive of response? Clin. Transl. Oncol. 2017, 19, 273–278. [Google Scholar] [CrossRef]
- Valdes-Rives, S.A.; Casique-Aguirre, D.; German-Castelan, L.; Velasco-Velazquez, M.A.; Gonzalez-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef] [Green Version]
- Hill, V.K.; Kim, J.S.; James, C.D.; Waldman, T. Correction of PTEN mutations in glioblastoma cell lines via AAV-mediated gene editing. PLoS ONE 2017, 12, e0176683. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, X.; Chen, L.; Du, W.; Cui, Y.; Piao, X.; Li, Y.; Jiang, C. Targeting glioma stem cells via the Hedgehog signaling pathway. Neuroimmunol. Neuroinflammation 2014, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Takezaki, T.; Hide, T.; Takanaga, H.; Nakamura, H.; Kuratsu, J.; Kondo, T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011, 102, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog and PI3K/Akt/mTOR pathways cooperate in suppressing survival, self-renewal and tumorigenic potential of glioblastoma-initiating cells. Mol. Cell. Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Chowdhary, S.A.; Ryken, T.; Newton, H.B. Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: A meta-analysis. J. Neurooncol. 2015, 122, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Song, A.; Andrews, D.W.; Werner-Wasik, M.; Kim, L.; Glass, J.; Bar-Ad, V.; Evans, J.J.; Farrell, C.J.; Judy, K.D.; Daskalakis, C.; et al. Phase I trial of alisertib with concurrent fractionated stereotactic re-irradiation for recurrent high grade gliomas. Radiother. Oncol 2019, 132, 135–141. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Huang, J.; Chaudhary, R.; Cohen, A.L.; Fink, K.; Goldlust, S.; Boockvar, J.; Chinnaiyan, P.; Wan, L.; Marcus, S.; Campian, J.L. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J. Neurooncol. 2019, 142, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Silvani, A.; De Simone, I.; Fregoni, V.; Biagioli, E.; Marchioni, E.; Caroli, M.; Salmaggi, A.; Pace, A.; Torri, V.; Gaviani, P.; et al. Multicenter, single arm, phase II trial on the efficacy of ortataxel in recurrent glioblastoma. J. Neurooncol. 2019, 142, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Ruda, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Lieberman, F.S.; Wang, M.; Robins, H.I.; Tsien, C.I.; Curran, W.J., Jr.; Werner-Wasik, M.; Smith, R.P.; Schultz, C.; Hartford, A.C.; Zhang, P.; et al. Phase 2 Study of Radiation Therapy Plus Low-Dose Temozolomide Followed by Temozolomide and Irinotecan for Glioblastoma: NRG Oncology RTOG Trial 0420. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 878–886. [Google Scholar] [CrossRef]
- Krauze, A.V.; Mackey, M.; Rowe, L.; Chang, M.G.; Holdford, D.J.; Cooley, T.; Shih, J.; Tofilon, P.J.; Camphausen, K. Late toxicity in long-term survivors from a phase 2 study of concurrent radiation therapy, temozolomide and valproic acid for newly diagnosed glioblastoma. Neurooncol. Pract. 2018, 5, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Gil-Gil, M.; Saran, F.; Carpentier, A.F.; Nowak, A.K.; Mason, W.; Zagonel, V.; Dubois, F.; Finocchiaro, G.; Fountzilas, G.; et al. A Randomized Phase II Trial (TAMIGA) Evaluating the Efficacy and Safety of Continuous Bevacizumab Through Multiple Lines of Treatment for Recurrent Glioblastoma. Oncologist 2019, 24, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; Fu, B.D.; Hsu, F.P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: Interim results and correlations with CD4(+) T-lymphocyte counts. CNS Oncol. 2018, 7, CNS22. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neurooncol. 2018, 140, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J.O.; Grossman, S.A.; Chi, A.S.; Mikkelsen, T.; Rosenfeld, M.R.; Ahluwalia, M.S.; Nabors, L.B.; Eichler, A.; Ribas, I.G.; Desideri, S.; et al. Phase II Study of Iniparib with Concurrent Chemoradiation in Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassman, A.B.; van den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Papadopoulos, K.P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro Oncol. 2019, 21, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, A.; Lamberti, G.; Cerbone, L.; Cordua, N.; Buonerba, C.; Peluso, G.; Di Lorenzo, G.; De Placido, S. High-dose fotemustine in temozolomide-pretreated glioblastoma multiforme patients: A phase I/II trial. Medicine (Baltimore) 2018, 97, e11254. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Li, J.; Boerner, J.; Stark, K.; Wu, J.; Kim, S.; Derogatis, A.; Mehta, S.; Dhruv, H.D.; Heilbrun, L.K.; et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin. Cancer Res. 2018, 24, 3820–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined With Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1195–1203. [Google Scholar] [CrossRef]
- Omuro, A.; Beal, K.; McNeill, K.; Young, R.J.; Thomas, A.; Lin, X.; Terziev, R.; Kaley, T.J.; DeAngelis, L.M.; Daras, M.; et al. Multicenter Phase IB Trial of Carboxyamidotriazole Orotate and Temozolomide for Recurrent and Newly Diagnosed Glioblastoma and Other Anaplastic Gliomas. J. Clin. Oncol. 2018, 36, 1702–1709. [Google Scholar] [CrossRef]
- Wirsching, H.G.; Tabatabai, G.; Roelcke, U.; Hottinger, A.F.; Jorger, F.; Schmid, A.; Plasswilm, L.; Schrimpf, D.; Mancao, C.; Capper, D.; et al. Bevacizumab plus hypofractionated radiotherapy versus radiotherapy alone in elderly patients with glioblastoma: The randomized, open-label, phase II ARTE trial. Ann. Oncol. 2018, 29, 1423–1430. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Natsume, A.; Mizusawa, J.; Katayama, H.; Fukuda, H.; Sumi, M.; Nishikawa, R.; Narita, Y.; Muragaki, Y.; Maruyama, T.; et al. JCOG0911 INTEGRA study: A randomized screening phase II trial of interferonbeta plus temozolomide in comparison with temozolomide alone for newly diagnosed glioblastoma. J. Neurooncol. 2018, 138, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Botero, G.; Cartalat-Carel, S.; Chinot, O.L.; Barrie, M.; Taillandier, L.; Beauchesne, P.; Catry-Thomas, I.; Barriere, J.; Guillamo, J.S.; Fabbro, M.; et al. Temozolomide Plus Bevacizumab in Elderly Patients with Newly Diagnosed Glioblastoma and Poor Performance Status: An ANOCEF Phase II Trial (ATAG). Oncologist 2018, 23, 524–e544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, D.; Jaeckle, K.A.; Anderson, S.K.; Galanis, E.; Giannini, C.; Buckner, J.C.; Stella, P.; Flynn, P.J.; Erickson, B.J.; Schwerkoske, J.F.; et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group Study/Alliance N0572. Cancer 2018, 124, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Lassman, A.B.; Schiff, D.; Yunus, S.A.; Gerstner, E.R.; Cloughesy, T.F.; Lee, E.Q.; Gaffey, S.C.; Barrs, J.; Bruno, J.; et al. Phase 2 and biomarker study of trebananib, an angiopoietin-blocking peptibody, with and without bevacizumab for patients with recurrent glioblastoma. Cancer 2018, 124, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neurooncol. 2018, 137, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E., 2nd; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157–e121. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.H.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro Oncol. 2018, 20, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Larsson, O.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501–81510. [Google Scholar] [CrossRef] [Green Version]
- Arrillaga-Romany, I.; Chi, A.S.; Allen, J.E.; Oster, W.; Wen, P.Y.; Batchelor, T.T. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget 2017, 8, 79298–79304. [Google Scholar] [CrossRef] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Ping, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients with prior antiangiogenic therapy. Neuro Oncol. 2018, 20, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Holland, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. 2018, 20, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Nghiemphu, P.L.; Ebiana, V.A.; Wen, P.; Gilbert, M.; Abrey, L.E.; Lieberman, F.; DeAngelis, L.M.; Robins, H.I.; Yung, W.K.A.; Chang, S.; et al. Phase I study of sorafenib and tipifarnib for recurrent glioblastoma: NABTC 05-02. J. Neurooncol. 2018, 136, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Du Four, S.; Bouttens, F.; Andre, C.; Verschaeve, V.; Van Fraeyenhove, F.; Chaskis, C.; D’Haene, N.; Le Mercier, M.; Rogiers, A.; et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J. Neurooncol. 2018, 136, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Badruddoja, M.A.; Pazzi, M.; Sanan, A.; Schroeder, K.; Kuzma, K.; Norton, T.; Scully, T.; Mahadevan, D.; Ahmadi, M.M. Phase II study of bi-weekly temozolomide plus bevacizumab for adult patients with recurrent glioblastoma. Cancer Chemother. Pharmacol. 2017, 80, 715–721. [Google Scholar] [CrossRef]
- Fariselli, L.; Cuppini, L.; Gaviani, P.; Marchetti, M.; Pinzi, V.; Milanesi, I.; Simonetti, G.; Tramacere, I.; DiMeco, F.; Salmaggi, A.; et al. Short course radiotherapy concomitant with temozolomide in GBM patients: A phase II study. Tumori 2017, 103, 457–463. [Google Scholar] [CrossRef]
- Yu, A.; Faiq, N.; Green, S.; Lai, A.; Green, R.; Hu, J.; Cloughesy, T.F.; Mellinghoff, I.; Nghiemphu, P.L. Report of safety of pulse dosing of lapatinib with temozolomide and radiation therapy for newly-diagnosed glioblastoma in a pilot phase II study. J. Neurooncol. 2017, 134, 357–362. [Google Scholar] [CrossRef]
- Sepulveda-Sanchez, J.M.; Vaz, M.A.; Balana, C.; Gil-Gil, M.; Reynes, G.; Gallego, O.; Martinez-Garcia, M.; Vicente, E.; Quindos, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 2017, 19, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Clarke, J.L.; Molinaro, A.M.; Cabrera, J.R.; DeSilva, A.A.; Rabbitt, J.E.; Prey, J.; Drummond, D.C.; Kim, J.; Noble, C.; Fitzgerald, J.B.; et al. A phase 1 trial of intravenous liposomal irinotecan in patients with recurrent high-grade glioma. Cancer Chemother. Pharmacol. 2017, 79, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Ursu, R.; Carpentier, A.; Metellus, P.; Lubrano, V.; Laigle-Donadey, F.; Capelle, L.; Guyotat, J.; Langlois, O.; Bauchet, L.; Desseaux, K.; et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma-A phase II multicentric, randomised study. Eur. J. Cancer 2017, 73, 30–37. [Google Scholar] [CrossRef]
- Nayak, L.; de Groot, J.; Wefel, J.S.; Cloughesy, T.F.; Lieberman, F.; Chang, S.M.; Omuro, A.; Drappatz, J.; Batchelor, T.T.; DeAngelis, L.M.; et al. Phase I trial of aflibercept (VEGF trap) with radiation therapy and concomitant and adjuvant temozolomide in patients with high-grade gliomas. J. Neurooncol. 2017, 132, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O(6)-Methylguanine-DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpathy-Cramer, J.; Chandra, V.; Da, X.; Ou, Y.; Emblem, K.E.; Muzikansky, A.; Cai, X.; Douw, L.; Evans, J.G.; Dietrich, J.; et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J. Neurooncol. 2017, 131, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Raizer, J.; Chamberlain, M.; Canelos, P.; Narwal, R.; Hong, S.; Miday, R.; Nade, M.; Laubscher, K. Phase II study of MEDI-575, an anti-platelet-derived growth factor-alpha antibody, in patients with recurrent glioblastoma. J. Neurooncol. 2017, 131, 185–191. [Google Scholar] [CrossRef]
- McCracken, D.J.; Celano, E.C.; Voloschin, A.D.; Read, W.L.; Olson, J.J. Phase I trial of dose-escalating metronomic temozolomide plus bevacizumab and bortezomib for patients with recurrent glioblastoma. J. Neurooncol. 2016, 130, 193–201. [Google Scholar] [CrossRef]
- Aoki, T.; Arakawa, Y.; Ueba, T.; Oda, M.; Nishida, N.; Akiyama, Y.; Tsukahara, T.; Iwasaki, K.; Mikuni, N.; Miyamoto, S. Phase I/II Study of Temozolomide Plus Nimustine Chemotherapy for Recurrent Malignant Gliomas: Kyoto Neuro-oncology Group. Neurol. Med. Chir. (Tokyo) 2017, 57, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Batchelor, T.T.; Gerstner, E.R.; Ye, X.; Desideri, S.; Duda, D.G.; Peereboom, D.; Lesser, G.J.; Chowdhary, S.; Wen, P.Y.; Grossman, S.; et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-beta tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol. 2017, 19, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Sautter, L.; Hofheinz, R.; Tuettenberg, J.; Grimm, M.; Vajkoczy, P.; Groden, C.; Schmieder, K.; Hochhaus, A.; Wenz, F.; Giordano, F.A. Open-Label Phase II Evaluation of Imatinib in Primary Inoperable or Incompletely Resected and Recurrent Glioblastoma. Oncology 2019, 98, 1–7. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Becker, K.P.; Mekhail, T.; Chowdhary, S.A.; Eakle, J.F.; Wright, D.; Langdon, R.M.; Yost, K.J.; Padula, G.D.A.; West-Osterfield, K.; et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J. Neurooncol. 2019, 144, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.J.; Panageas, K.S.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neurooncol. 2019, 144, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Schilero, C.; Peereboom, D.M.; Hobbs, B.P.; Elson, P.; Stevens, G.H.J.; McCrae, K.; Nixon, A.B.; Ahluwalia, M.S. Phase II study of Dovitinib in recurrent glioblastoma. J. Neurooncol. 2019, 144, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Du, X.J.; Li, X.M.; Cai, L.B.; Sun, J.C.; Wang, S.Y.; Wang, X.C.; Pang, X.L.; Deng, M.L.; Chen, F.F.; Wang, Z.Q.; et al. Efficacy and safety of nimotuzumab in addition to radiotherapy and temozolomide for cerebral glioblastoma: A phase II multicenter clinical trial. J. Cancer 2019, 10, 3214–3223. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Q.; Muzikansky, A.; Duda, D.G.; Gaffey, S.; Dietrich, J.; Nayak, L.; Chukwueke, U.N.; Beroukhim, R.; Doherty, L.; Laub, C.K.; et al. Phase II trial of ponatinib in patients with bevacizumab-refractory glioblastoma. Cancer Med. 2019, 8, 5988–5994. [Google Scholar] [CrossRef]
- Weller, J.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Hau, P.; Krex, D.; Grauer, O.; Goldbrunner, R.; Bahr, O.; et al. Health-related quality of life and neurocognitive functioning with lomustine-temozolomide versus temozolomide in patients with newly diagnosed, MGMT-methylated glioblastoma (CeTeG/NOA-09): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1444–1453. [Google Scholar] [CrossRef]
- Lapointe, S.; Mason, W.; MacNeil, M.; Harlos, C.; Tsang, R.; Sederias, J.; Luchman, H.A.; Weiss, S.; Rossiter, J.P.; Tu, D.; et al. A phase I study of vistusertib (dual mTORC1/2 inhibitor) in patients with previously treated glioblastoma multiforme: A CCTG study. Investig. New Drugs 2019. [Google Scholar] [CrossRef]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage Exclusion after Radiation Therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef] [Green Version]
- Van den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.S.; Franceschi, E.; Clement, P.M.; Chinot, O.; de Vos, F.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFRamplified glioblastoma. Neuro Oncol. 2019. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Brenner, A.; de Groot, J.F.; Butowski, N.A.; Zach, L.; Campian, J.L.; Ellingson, B.M.; Freedman, L.S.; Cohen, Y.C.; Lowenton-Spier, N.; et al. A randomized controlled phase III study of VB-111 combined with bevacizumab vs. bevacizumab monotherapy in patients with recurrent glioblastoma (GLOBE). Neuro Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.A.; Friedman, D.D.; Akbari, S.H.A.; Kim, A.H.; Tao, Y.; Luo, J.; Leuthardt, E.C. Glioblastoma Treated With Magnetic Resonance Imaging-Guided Laser Interstitial Thermal Therapy: Safety, Efficacy, and Outcomes. Neurosurgery 2019, 84, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoudi, K.; Bouras, A.; Bozec, D.; Ivkov, R.; Hadjipanayis, C. Magnetic hyperthermia therapy for the treatment of glioblastoma: A review of the therapy’s history, efficacy and application in humans. Int. J. Hyperth. 2018, 34, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuthardt, E.C.; Duan, C.; Kim, M.J.; Campian, J.L.; Kim, A.H.; Miller-Thomas, M.M.; Shimony, J.S.; Tran, D.D. Hyperthermic Laser Ablation of Recurrent Glioblastoma Leads to Temporary Disruption of the Peritumoral Blood Brain Barrier. PLoS ONE 2016, 11, e0148613. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Patel, N.V.; Danish, S.F. Intracranial MR-guided laser-induced thermal therapy: Single-center experience with the Visualase thermal therapy system. J. Neurosurg. 2016, 125, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.G.; Rao, G.; Kew, Y.; Prabhu, S.S. Laser interstitial thermal therapy for newly diagnosed and recurrent glioblastoma. Neurosurg. Focus 2016, 41, E12. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, A.M.; Hawasli, A.H.; Rodriguez, A.; Schroeder, J.L.; Laxton, A.W.; Elson, P.; Tatter, S.B.; Barnett, G.H.; Leuthardt, E.C. The role of laser interstitial thermal therapy in enhancing progression-free survival of difficult-to-access high-grade gliomas: A multicenter study. Cancer Med. 2014, 3, 971–979. [Google Scholar] [CrossRef]
- Davies, A.M.; Weinberg, U.; Palti, Y. Tumor treating fields: A new frontier in cancer therapy. Ann. N. Y. Acad. Sci. 2013, 1291, 86–95. [Google Scholar] [CrossRef]
- Rick, J.; Chandra, A.; Aghi, M.K. Tumor treating fields: A new approach to glioblastoma therapy. J. Neurooncol. 2018, 137, 447–453. [Google Scholar] [CrossRef]
- Optune® Elevate Expectations. INSTRUCTIONS FOR USE. Available online: https://www.optune.com/Content/pdfs/Optune_IFU_8.5x11.pdf (accessed on 10 March 2020).
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Taphoorn, M.J.B.; Dirven, L.; Kanner, A.A.; Lavy-Shahaf, G.; Weinberg, U.; Taillibert, S.; Toms, S.A.; Honnorat, J.; Chen, T.C.; Sroubek, J.; et al. Influence of Treatment With Tumor-Treating Fields on Health-Related Quality of Life of Patients With Newly Diagnosed Glioblastoma: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 495–504. [Google Scholar] [CrossRef]
- Desjardins, A.; Vlahovic, G.; Friedman, H.S. Vaccine Therapy, Oncolytic Viruses, and Gliomas. Oncology (Williston Park) 2016, 30, 211–218. [Google Scholar] [PubMed]
- Tivnan, A.; Heilinger, T.; Lavelle, E.C.; Prehn, J.H. Advances in immunotherapy for the treatment of glioblastoma. J. Neurooncol. 2017, 131, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro Oncol. 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers (Basel) 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers (Basel) 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Sayegh, E.T.; Oh, T.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Vaccine therapies for patients with glioblastoma. J. Neurooncol. 2014, 119, 531–546. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Options Oncol. 2019, 20, 24. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Treatment | Disease Type | Clinical Trial Phase | No. of Patients | Result (s) | Reference |
|---|---|---|---|---|---|
| 1 Alisertib + RT | Recurrent | I | 17 | OS-6: 88.2%; Median survival: 11.1 months; PFS-6: 35.5%. | [84] |
| 2 Lomustine + 52 TMZ vs. 52TMZ | Primary | III | 141 | Median OS: 48.1 months (32.6—not assessable) vs. 31.4 months (95% CI, 27.7—47.1); AEs: 59% vs. 51% of patients. | [85] |
| 3 Disulfiram + copper | Recurrent | II | 21 | ORR: 0%; Clinical benefit: 14%; Median PFS: 1.7 months; Median OS: 7.1 months; DLTs: 4%. | [86] |
| 4 Ortataxel | Recurrent | II | 40 | PFS-6: 11.4%; AEs: Neutropenia and hepatotoxicity (13.2%) and leukopenia (15.8%). | [87] |
| 5 Buparlisib | Recurrent | II | 15+50 | Reduction of phosphorylated AKT: 42.8%; PFS-6: 8%; Median PFS: 1.7 months (95% CI, 1.4 to 1.8 months); AEs: Lipase elevation (10.8%), fatigue (6.2%), hyperglycemia (4.6%), elevated ALT (4.6%). | [88] |
| 6 Regorafenib vs. 2 Lomustine | Recurrent | II | 119 | Patients died at cut-off: 71% vs 95%; Median OS: 7.4 months (95% CI, 5.8—12.0) vs. 5.6 months (95% CI, 4.7–7.3); AEs: 56% (hand–foot skin reaction, increased lipase, blood bilirubin) vs. 40% (decreased platelet count, decreased lymphocyte count, neutropenia). | [89] |
| 52 TMZ + RT → 52 TMZ + 7 irinotecan (CPT-11) | Primary | II | 152 | Median OS: 16.9 months vs 13. 7 months (p = 0.03) in historical control; Grade 3/4 hematologic toxicity: 38% vs. 14% in Stupp trial. | [90] |
| 8 Valproic acid + 52 TMZ + RT | Primary | II | 6 | Late toxicity in long-term survivors: neurological, pain, and blood/bone marrow toxicity (mostly grade 1/2). | [91] |
| 52 TMZ + 9 memantine + 10 mefloquine + 11 metformin (adjuvant) | Primary | I | 81 | DLTs: Dizziness (memantine), gastrointestinal effects (metformin); AEs: Lymphopenia (66%); Median survival: 21 months; 2-year survival: 43%; MTDs (doublet, triplet, quadruplet): Memantine (20 mg b.i.d., 10 mg b.i.d., 10 mg b.i.d.), mefloquine (250 mg 3 times weekly, 250 mg 3 times weekly, 250 mg 3 times weekly), metformin (850 mg b.i.d., 850 mg b.i.d., 500 mg b.i.d.). | [92] |
| RT + 52 TMZ + 12 bevacizumab (BEV) → 2 CCNU + 12 BEV/ 2 CCNU + placebo → 12 BEV/placebo + chemotherapy | Recurrent | II | 296 | No survival benefit and no safety concerns. | [93] |
| 13 ERC1671 + 12 bevacizumab vs. 12 bevacizumab + placebo | Recurrent | II | 9 | Median OS: 12 months vs. 7.5 months. | [94] |
| 14 Palbociclib (with and without resection) | Recurrent | II | 22 | Median PFS: 5.14 weeks (5 days–142 weeks); Median OS: 15.4 weeks (2–274 weeks). | [95] |
| 15 Iniparib + RT + 52 TMZ | Primary | II | 81 | Median OS: 22 months (95% CI, 17-24); 2- and 3-year survival: 38% and 25%; Grade 3 AEs: 27% of patients. | [96] |
| 16 Depatuxizumab mafodotin + 52 TMZ | Recurrent | I | 60 | AEs: blurred vision (63%), fatigue (38%), and photophobia (35%); Grade 3/4 AEs: Ocular (22%), non-ocular (22%); ORR: 14.3%; PFS-6: 25.2%; OS-6: 69.1%. | [97] |
| 17 Fotemustine (120 or 140 mg/m) | Recurrent | I/II | 37 | Toxicity: Grade 3 and 4 thrombocytopenia (4 of 6 patients at 140 mg/m vs. 3 of 31 patients at 120 mg/m); Median PFS: 12.1 (1–40.2) weeks; OS: 19.7 (1–102) weeks. | [98] |
| 18 AZD1775 | Recurrent | 0 | 20 | BBB permeability; Median unbound tumor to plasma concentration ratio: 3.2. | [99] |
| 19 Bortezomib + 52 TMZ + RT | Primary | II | 24 | Median PFS: 6.2 months (95% CI 3.7–8.8); Median OS: 19.1 months (95% CI, 6.7–31.4); no unexpected AEs. | [100] |
| 20 Carboxyam- idotriazole orotate + 52TMZ | Recurrent/ Primary | Ib | 47 | DLTs: none; Recommended phase II dose: 600 mg/day. | [101] |
| 12 Bevacizumab + RT vs. RT | Primary | II | 75 | Median PFS: 7.6 vs. 4.8 months, p = 0.003; OS: 12.1 vs. 12.2 months, p = 0.77. | [102] |
| 21 Interferon β + 52 TMZ + RT vs. 52TMZ + RT | Primary | II | 122 | OS: 24.0 vs. 20.3 months; Median PFS: 8.5 vs. 10.1 months; Neutropenia: 20.7 vs. 12.7 % (concomitant) and 9.3% vs. 3.6% (maintenance). | [103] |
| 12 Bevacizumab + 52 TMZ | Primary | II | 66 | Median OS: 23.9 weeks (95% CI 19–27.6); Median PFS: 15.3 weeks (95% CI, 12.9–19.3); AEs: Grade ≥ 3 hematological events (20%), high blood pressure (24%), venous thromboembolism (4.5%), cerebral hemorrhage (3%), and Intestinal perforation (3%). | [104] |
| 3 Disulfiram (with or without copper) + adjuvant 52 TMZ | Primary | I | 18 | MTD: Disulfiram 500 mg daily was well tolerated, 1000 mg daily was not; Median PFS: 4.5 months (95% CI 0.8–8.2); Median OS: 14.0 months (95% CI 8.3–19.6). | [7] |
| 22 Temsirolimus + 23 sorafenib | Recurrent | I/II | 41 | MTD (Phase I): sorafenib (200 mg twice daily) and Temsirolimus (20 mg weekly); Median PFS and OS (Phase II): 2.6 months vs. 1.9 months (VEGF inhibitor-naïve vs. prior VEGF inhibitor patients) and 6.3 months vs. 3.9 months (VEGF inhibitor-naïve vs. prior VEGF inhibitor patients). | [105] |
| 24 Trebananib vs. 24 trebananib + 12 bevacizumab | Recurrent | II | 48 | Trebananib: Well tolerated as monotherapy; Trebananib + Bevacizumab: PFS-6 (24.3%, 95% CI, 12.1%-38.8%), Median OS (9.5 months, 95% CI, 7.5–4.7 months), OS-12 (37.8%, 95% CI, 22.6%–53.0%). | [106] |
| 25 Vorinostat + 12 bevacizumab + 52 TMZ | Recurrent | I/II | 9+39 | MTD (phase I): 400 mg for vorinostat; PFS-6 (phase II): 53.8% (95% CI, 37.2–67.9). | [107] |
| 25 Vorinostat + 12 bevacizumab | Recurrent | II | 40 | PFS-6: 30.0% (95% CI, 16.8%–44.4%); Median OS: 10.4 months (95% CI, 7.6–12.8 months); AEs (grade 2): Lymphopenia (55%), leukopenia (45%), neutropenia (35%), and hypertension (33%). AEs (grade 4): Leukopenia (3%), neutropenia (3%), sinus bradycardia (3%), and venous thromboembolism (3%). | [108] |
| 26 Everolimus + RT + 52 TMZ vs. RT + 52TMZ | Primary | II | 171 | Median PFS: 8.2 vs. 10.2 months, p = 0.79); Median OS: 16.5 vs. 21.2 months, p = 0.008) | [109] |
| 27 AXL1717 | Recurrent | I | 9 | Tumor response: 44%; AEs: Neutropenia. | [110] |
| 28 ONC201 | Recurrent | II | 17 | Median OS: 41.6 weeks; PFS-6: 11.8%; Drug-related serious AEs: None; Plasma pharmacokinetics (2-h post-dose): 2.6 µg/mL. | [111] |
| 29 Nivolumab (with or without 30 ipilimumab) | Recurrent | I | 40 | Nivolumab monotherapy better tolerated; AEs: fatigue, and diarrhea; Tumor-cell programmed death ligand-1 expression ≥1% (68%). | [112] |
| 31 Cabozantinib | Recurrent | II | 70 | ORR: 4.3%; Median duration of response: 4.2 months; PFS-6: 8.5%; Median PFS: 2.3 months; Median OS: 4.6 months. AEs: Fatigue, diarrhea, increased alanine aminotransferase, headache, hypertension, and nausea. 48.6% resulted in dose reductions (140 mg/day to 100 mg/day). | [113] |
| 31 Cabozantinib (140 mg/day vs. 100 mg/day) | Recurrent | II | 152 | ORR: 17.6% vs. 14.5%; PFS-6: 22.3% vs. 27.8%; Median PFS: 3.7 months in both; Median OS: 7.7 vs. 10.4 months; AEs (grade 3/4): 79.4% vs. 84.7%; Dose reduction due to AEs: 61.8% vs. 72.0%. | [114] |
| 25 Vorinostat + 52 TMZ + RT | Primary | I/II | 15+107 | MTD: 300 mg/day; DLTs: Grade 4 neutropenia and thrombocytopenia and grade 3 aspartate aminotransferase elevation, hyperglycemia, fatigue, and wound dehiscence; Phase II OS-15 months: 55.1% (median OS 16.1 month); Phase II toxicities: Lymphopenia (32.7%), thrombocytopenia (28.0%), and neutropenia (21.5%). | [115] |
| 23 Sorafenib + 32 tipifarnib | Recurrent | I | 24 | Study stopped because of excessive toxicities. Last dose reached: 200 mg and 100 mg twice a day for sorafenib and tipifarnib, respectively. | [116] |
| 33 Axitinib vs. 33 axitinib + 2 lomustine | Recurrent | II | 79 | ORR: 28% vs. 38%; PFS-6: 26% (95% CI, 14–38) vs. 17% (95% CI, 2–32); Median OS: 29 weeks (95% CI, 20–38) vs. 27.4 weeks (95% CI 18.4–36.5); Toxicities: Grade ¾ neutropenia (0 vs. 21%) and thrombocytopenia (4 vs. 29%). | [117] |
| 34 Rindopepimut + 52 TMZ vs. 52 TMZ | Primary | III | 745 | OS for patients with MRD: 20.1 months (95% CI, 18.5–22.1) vs. 20.0 months (18.1–21.9); Grade 3/4 AEs: Thrombocytopenia (9% vs. 6%), fatigue (2% vs. 5%), brain edema (2% vs. 3%), seizure (2% vs. 2%), and headache (2% vs. 3%); Mortality by AEs: 4% vs. 3%. | [118] |
| 12 Bevacizumab + 52 TMZ | Recurrent | II | 30 | ORR: 51 weeks; PFS-6: 52%; Median time to tumor progression: 5.5 months. | [119] |
| 52 TMZ (150–200 mg/m2/day) + RT (60 Gy in 5 days) | Primary | II | 35 | OS: 22 months; Hematologic toxicities: ≤grade 2. | [120] |
| 35 Lapatinib + 52TMZ + RT | Primary | II | 12 | Higher dose correlates to lymphopenia; Common AEs: fatigue, rashes, and diarrhea | [121] |
| 36 Dacomitinib | Recurrent | II | 30 + 19 | PFS-6: 10.6%; Median PFS: 2.7 months; Median OS: 7.4; Best overall response: 4.1%; Common AEs: Diarrhea and rash; Drug-related AEs: 40.8% (grade 3/4). | [122] |
| 37 HER2-CAR VSTs (HER2 specific CAR-modified virus-specific T cells) | Recurrent | I | 17+7 | No dose-limiting toxic effects; Presence in peripheral blood: up to 12 months; Stable disease: 7 out of 16 patients for 8 weeks to 29 months; Disease progression: 8 out of 16 patients; Median OS: 11.1 months (95% CI, 4.1–27.2 months) after infusion. | [123] |
| 38 Irinotecan liposome injection (nal-IRI) | Recurrent | I | 16 + 18 | MTD: 120 mg/m2 (WT cohort), 150 mg/m2 (HT cohort); DLTs: Diarrhea, dehydration and/or fatigue. | [124] |
| 39 CpGODN→RT + 52 TMZ vs. RT + 52 TMZ | Primary | II | 81 | 2 years OS: 31% vs. 26%; Median PFS: 9 vs. 8.5 months. | [125] |
| 40 Aflibercept + RT + 52 TMZ→ 52 TMZ | Primary | I | 59 | MTD: 4 mg/kg for 2 weeks; DLTs: G3 deep vein thrombosis, G4 neutropenia, G4 biopsy-confirmed thrombotic microangiopathy, G3 rash, G4 thrombocytopenia; Treatment discontinuation: disease progression (47%), toxicities (36%), others (14%), full course (3%). | [126] |
| 41 Onartuzumab + 12 bevacizumab vs. placebo + 12 bevacizumab | Recurrent | II | 129 | Median PFS: 3.9 vs. 2.9 months; Median OS: 8.8 vs. 12.6 months; AEs (G ≥ 3): 38.5% vs. 35.9%. | [127] |
| 42 Tivozanib | Recurrent | II | 10 | Progressive disease: 80%; Median PFS: 2.3 months; Median OS: 8.1 months. | [128] |
| 43 MEDI-575 | Recurrent | II | 56 | PFS-6: 15.4% (90% CI 8.1–24.9 months); Stable disease: 41.1%; Median PFS: 1.4 months (90% CI 1.4–1.8); Median OS: 9.7 months (90% CI, 6.5–11.8); Treatment-related AEs: Diarrhea (16%), nausea (13%), and fatigue (13%). | [129] |
| 44 Bortezomib + 52 TMZ + 12 bevacizumab | Recurrent | I | 12 | MTD: 75 mg/m2 for TMZ; PFS: 3.27 months: Mean OS: 20.75 months. | [130] |
| 45 Nimustine + 52 TMZ | Recurrent | I/II | 15 + 40 | MTD: TMZ (150 mg/m2), nimustine (40 mg/m2); ORS: 11%; Stable disease: 68%; PFS-6 and PFS-12: 24% (95% CI, 12–35%) and 8% (95% CI, 4–15%); Median PFS: 13 months (95% CI, 9.2–17.2 months); OS-6 and OS-12: 78% (95% CI, 67–89%) and 49% (95% CI, 33–57%); Median OS: 11.8 months (95% CI, 8.2–14.5 months). | [131] |
| 46 Tandutinib | Recurrent | I/II | 19+30 | MTD: 600 mg twice daily; Phase II terminated as PFS-6 not achieved. | [132] |
| 47 Imatinib + RT vs. 47 imatinib + re-irradation | Recurrent | II | 51 | Median OS: 5.0 months (95% CI, 0-24.1 months) vs. 6.5 months (95% CI 0–32.5 months; Median PFS: 2.8 months (95% CI 0–8.7 months) vs. 2.1 months (95% CI 0–11.8 months). | [133] |
| 48 BKM120 + 12 bevacizumab | Recurrent | I/II | 88 | MTD: 60 mg PO (orally) daily; PFS-6: 36.5%; ORR: 26%; TRTs: 57%. | [134] |
| 49 Perifosine | Recurrent | II | 30 | PFS-6: 0%; PFS: 1.58 months (95% CI, 1.08–1.84 months); Median OS: 3.68 months (95% CI, 2.50–7.79 months). | [135] |
| 50 Dovitinib (naïve vs. progressed on prior antiangiogenic therapy) | Recurrent | II | 19+14 | PFS-6: 12% vs. 0%; TTP: median 1.8 months vs. 0.7–1.8 months. | [136] |
| 51 Nimotuzumab + 52 TMZ + RT | Primary | II | 39 | ORR: 72.2%; Median OS: 24.5 months; Median PFS: 11.9 months. | [137] |
| 53 Ponatinib | Recurrent | II | 15 | PFS-3: 0; Median PFS: 28 days (95% CI, 27–30); Median OS: 98 days (95% CI 56–257). | [138] |
| 2 Lomustine + 52 TMZ vs. 52 TMZ | Primary | III | 129 | Health-related quality of life: No significant differences; Neurocognitive function: Mini-mental state examination (favors the TMZ group); Neurocognitive test battery: No significant differences. | [139] |
| 54Vistusertib + 52TMZ | Recurrent | I | 15 | Tolerability: Vistusertib 125 mg b.i.d. + TMZ 150 mg/m2 for 5 days; PFS-6: 26.6%; AEs: G1/G2. | [140] |
| 55 Ascorbate + RT + 52 TMZ | Primary | I | 11 | DLTs: None; AEs: Dry mouth and chills; Median PFS: 9.4 months; Median OS: 18 months. | [141] |
| 56 Plerixafor | Primary | I/II | 9+20 | Tolerability: No drug-attributable G3 toxicities; Median OS: 21.3 months (95% CI, 15.9-NA); PFS: 14.5 months (95% CI, 11.9-NA). | [142] |
| 16 Depatux-M (+52 TMZ) vs. 52 TMZ/ 2 lomustine | Recurrent | II | 260 | Efficacy: Monotherapy is comparable to control (hazard ratio: 1.04, 95% CI, 0.73–1.48); Toxicities: Reversible corneal epitheliopathy; AEs: G3–G4 (25–30%) | [143] |
| 57 VB-111 + 12 bevacizumab vs. 12 bevacizumab | Recurrent | III | 256 | Median OS: 6.8 months (combination) vs. 7.9 months (control); ORR: 27.3% (combination) vs. 21.9% (control); AEs (G3–G5): 67% (combination) vs. 40% (control). | [144] |
| Therapeutic Agent | Disease Type | Dosage Regimen |
|---|---|---|
| Temozolomide (TMZ) | Newly diagnosed | Concurrent: 75 mg/m2 daily for six weeks with focal RT. Adjuvant *: Starts followed by a 4-week rest period after concurrent therapy. 1st cycle, 150 mg/m2 daily for five days in a 28-day cycle. 150–200 mg/m2 daily for 5 days in a 28-day cycle, 2nd–6th cycles. |
| Bevacizumab | Recurrent | 10 mg/kg as intravenous infusion every 2 weeks **. |
| Carmustine (BiCNU) for injection | - | 150–200 mg/m2 (single or divided into two successive days) intravenously every 6 weeks. |
| Carmustine (BiCNU) implant | Newly diagnosed/Recurrent | Eight 7.7 mg wafers with a total of 61.6 mg implanted intracranially. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajaratnam, V.; Islam, M.M.; Yang, M.; Slaby, R.; Ramirez, H.M.; Mirza, S.P. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers 2020, 12, 937. https://doi.org/10.3390/cancers12040937
Rajaratnam V, Islam MM, Yang M, Slaby R, Ramirez HM, Mirza SP. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers. 2020; 12(4):937. https://doi.org/10.3390/cancers12040937
Chicago/Turabian StyleRajaratnam, Vilashini, Mohammad Mohiminul Islam, Maixee Yang, Rachel Slaby, Hilda Martinez Ramirez, and Shama Parveen Mirza. 2020. "Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments" Cancers 12, no. 4: 937. https://doi.org/10.3390/cancers12040937