Landscape of Mitochondria Genome and Clinical Outcomes in Stage 1 Lung Adenocarcinoma

 , , add
Show full author list
, , add
Show full author list
Abstract
:1. Introduction
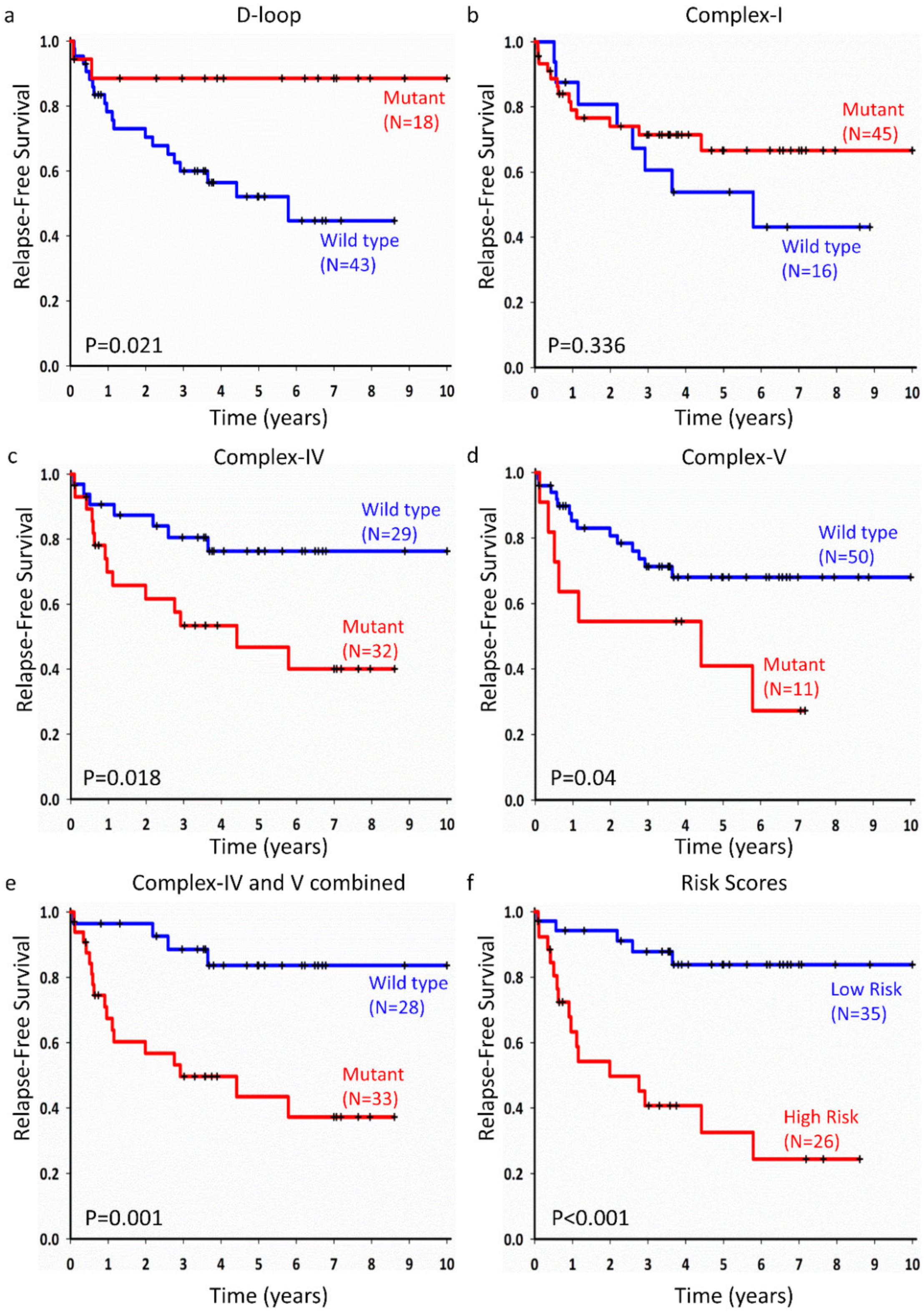
2. Results
2.1. Clinical and Demographic Characteristics of Study Cohort
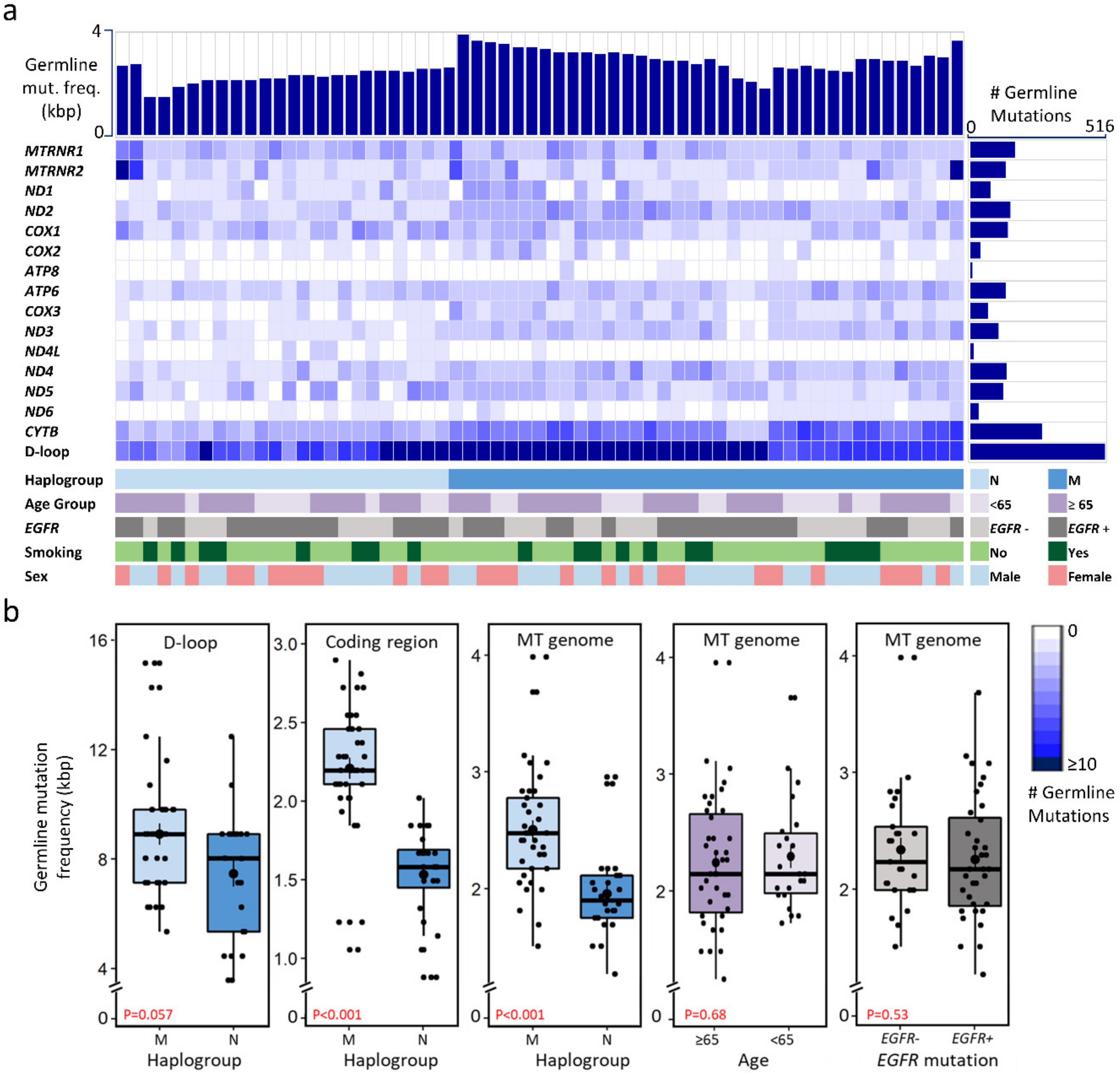
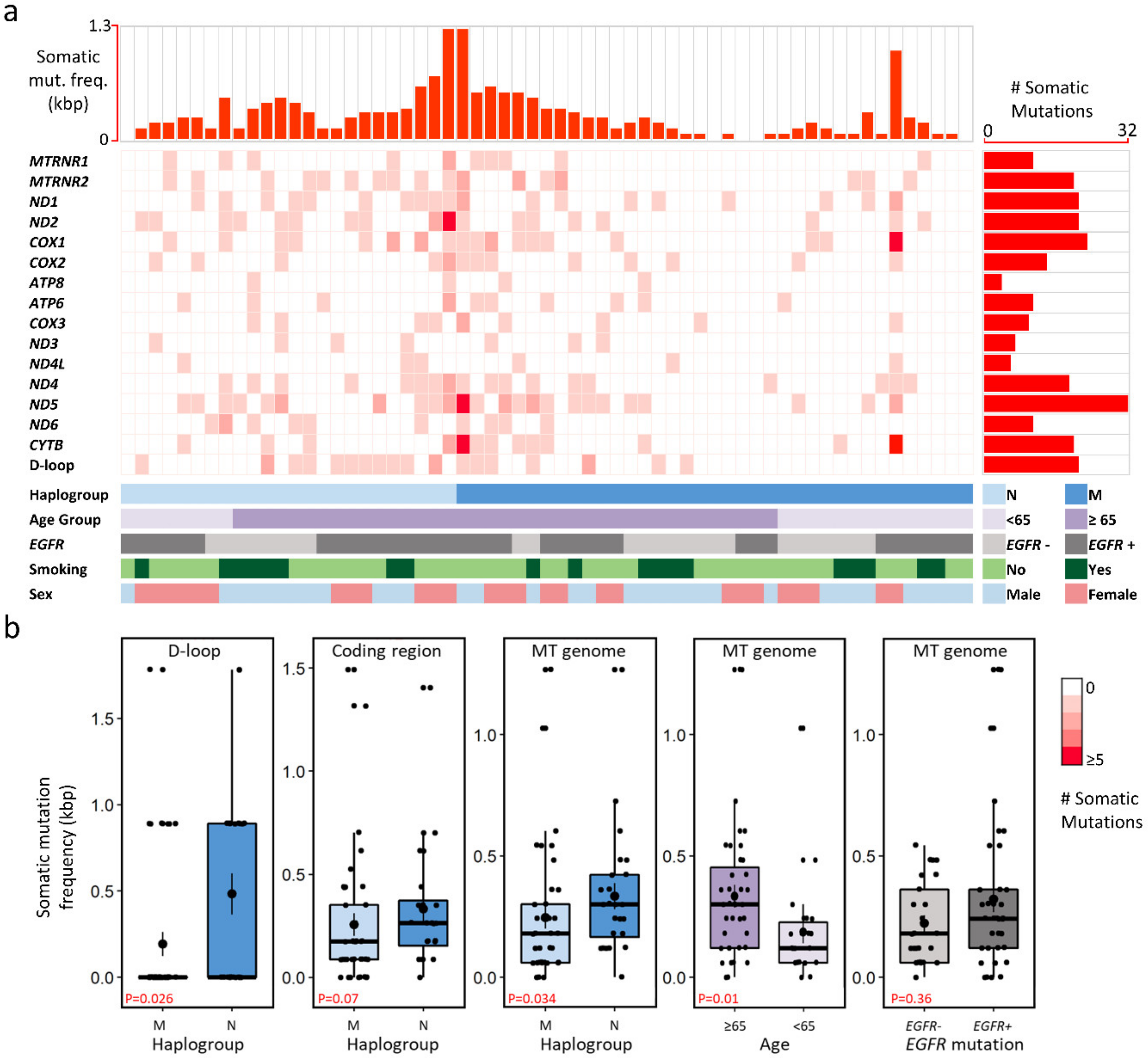
2.2. Mitochondrial Mutational Spectrum
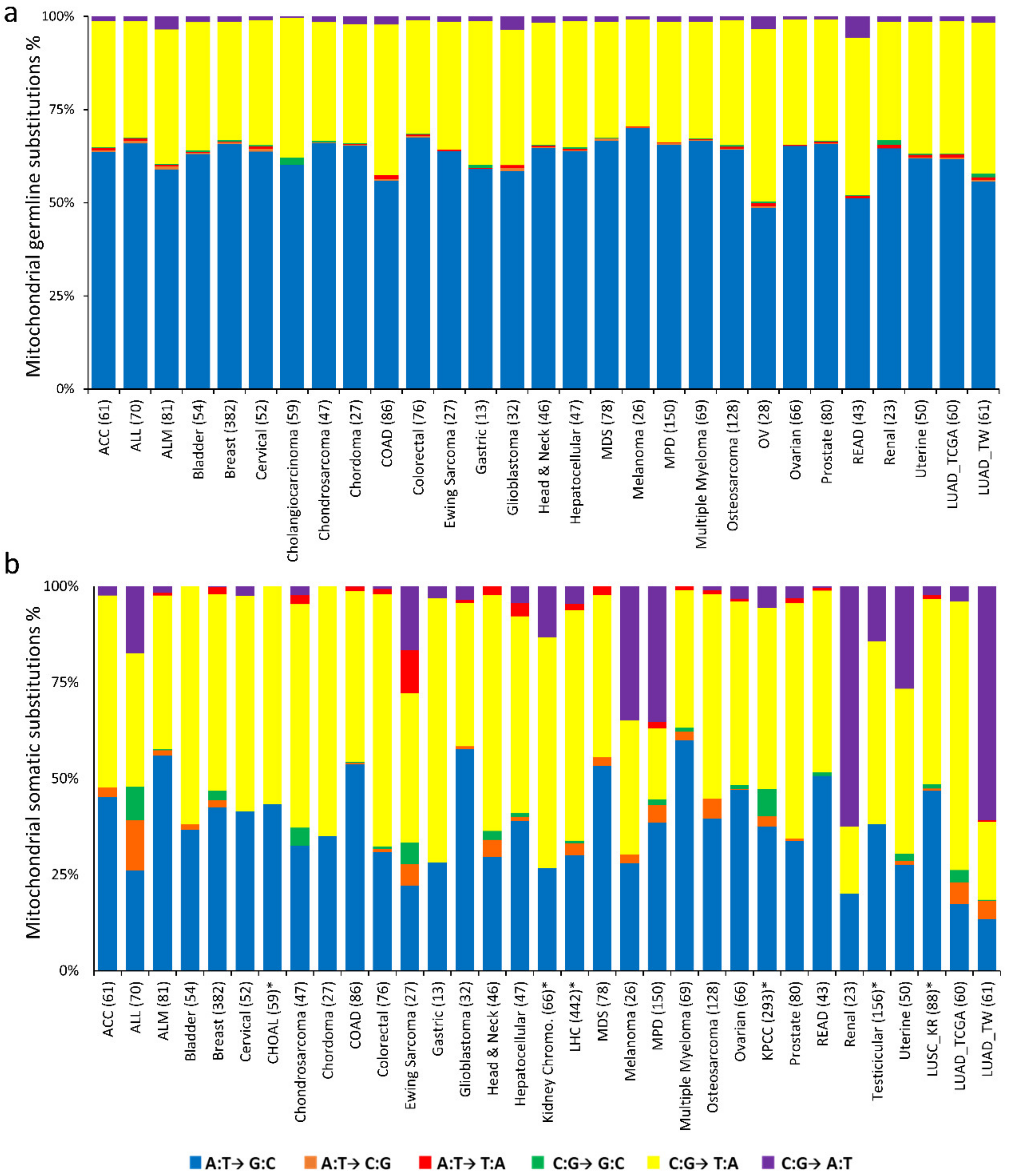
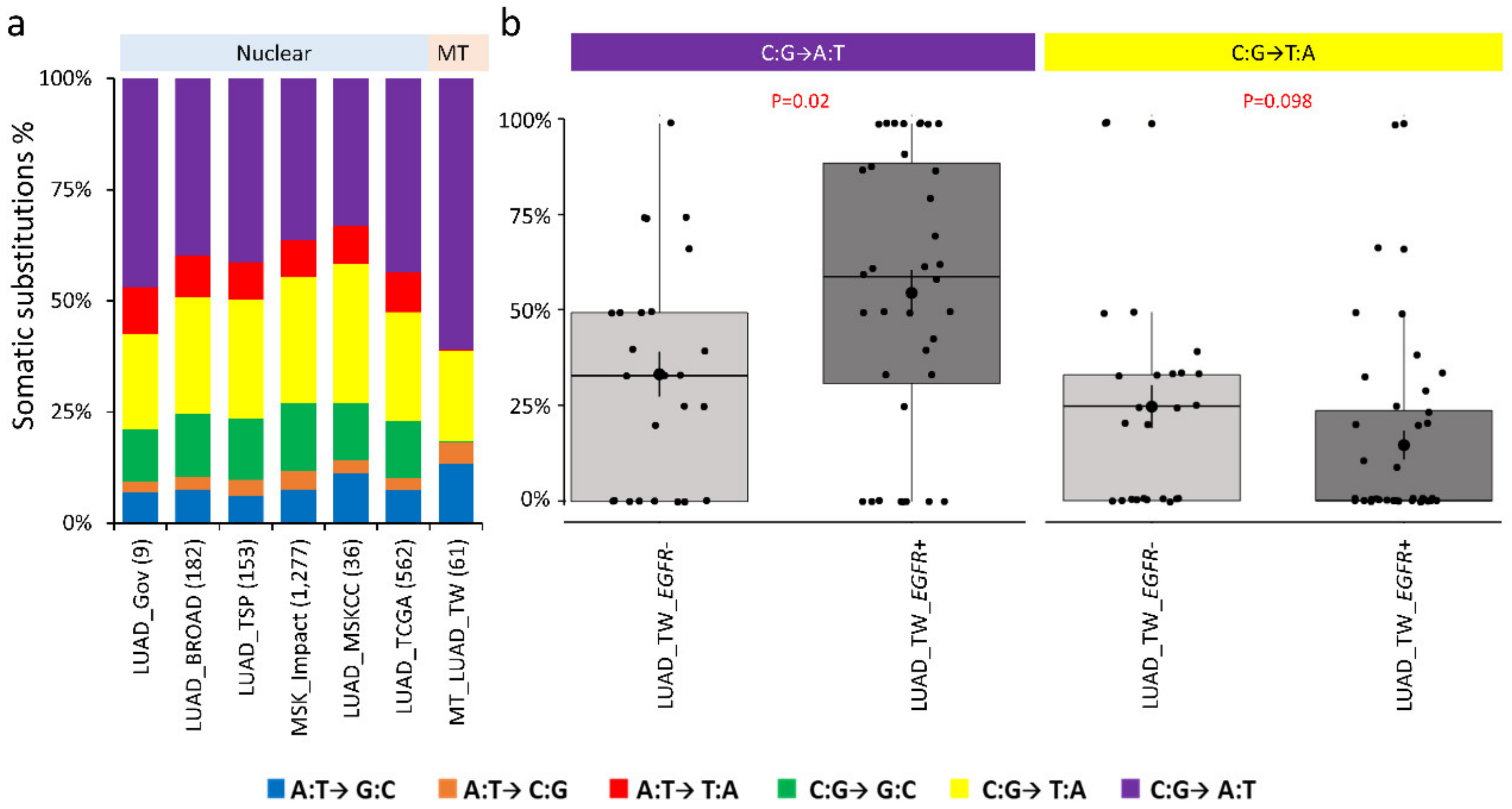
2.3. Nucleotide Substitution Profiles of Mitochondrial Mutations
2.4. Mitochondrial Somatic Mutations and Prognosis of Lung Adenocarcinoma
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Mitochondrial Isolation, DNA Extraction, and Sequencing
4.3. EGFR Genotyping
4.4. Bioinformatics Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Swanton, C.; Govindan, R. Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Ost, D.; Goldberg, J.; Rolnitzky, L.; Rom, W.N. Survival after surgery in stage ia and ib non-small cell lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sazanov, L.A.; Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex i from thermus thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.A.; Doda, J.N.; Friedberg, E.C. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 1974, 71, 2777–2781. [Google Scholar] [CrossRef] [Green Version]
- Royal, C.D.; Novembre, J.; Fullerton, S.M.; Goldstein, D.B.; Long, J.C.; Bamshad, M.J.; Clark, A.G. Inferring genetic ancestry: Opportunities, challenges, and implications. Am. J. Hum. Genet. 2010, 86, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial-DNA sequence variation in human-evolution and disease. Proc. Natl. Acad. Sci. USA 1994, 91, 8739–8746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.Z.; Shen, L.J.; Chen, T.; He, J.; Ding, Z.N.; Wei, J.; Qu, J.C.; Chen, G.R.; Lu, J.X.; Bai, Y.D. Cancer type-specific modulation of mitochondrial haplogroups in breast, colorectal and thyroid cancer. BMC Cancer 2010, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.K.; Leal, S.M.; Covarrubias, D.; Liu, A.; Wong, L.J.C. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007, 67, 4687–4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triska, P.; Kaneva, K.; Merkurjev, D.; Sohail, N.; Falk, M.J.; Triche, T.J.; Biegel, J.A.; Gai, X.W. Landscape of germline and somatic mitochondrial DNA mutations in pediatric malignancies. Cancer Res. 2019, 79, 1318–1330. [Google Scholar] [CrossRef] [Green Version]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [Green Version]
- Hertweck, K.L.; Dasgupta, S. The landscape of mtdna modifications in cancer: A tale of two cities. Front. Oncol. 2017, 7, 262. [Google Scholar] [CrossRef]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial reprogramming regulates breast cancer progression. Clin. Cancer Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005, 65, 1655–1663. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [Green Version]
- Lievre, A.; Blons, H.; Houllier, A.M.; Laccourreye, O.; Brasnu, D.; Beaune, P.; Laurent-Puig, P. Clinicopathological significance of mitochondrial d-loop mutations in head and neck carcinoma. Br. J. Cancer 2006, 94, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Tommasi, S.; Favia, P.; Weigl, S.; Bianco, A.; Pilato, B.; Russo, L.; Paradiso, A.; Petruzzella, V. Mitochondrial DNA variants and risk of familial breast cancer: An exploratory study. Int. J. Oncol. 2014, 44, 1691–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaffarpour, M.; Mahdian, R.; Fereidooni, F.; Kamalidehghan, B.; Moazami, N.; Houshmand, M. The mitochondrial atpase6 gene is more susceptible to mutation than the atpase8 gene in breast cancer patients. Cancer Cell Int. 2014, 14, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazdal, D.; Harms, A.; Endris, V.; Penzel, R.; Kriegsmann, M.; Eichhorn, F.; Muley, T.; Stenzinger, A.; Pfarr, N.; Weichert, W.; et al. Prevalence of somatic mitochondrial mutations and spatial distribution of mitochondria in non-small cell lung cancer. Br. J. Cancer 2017, 117, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Soudry, E.; Mukhopadhyay, N.; Shao, C.; Yee, J.; Lam, S.; Lam, W.; Zhang, W.; Gazdar, A.F.; Fisher, P.B.; et al. Mitochondrial DNA mutations in respiratory complex-i in never-smoker lung cancer patients contribute to lung cancer progression and associated with egfr gene mutation. J. Cell. Physiol. 2012, 227, 2451–2460. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Cairns, R.; Papandreou, I.; Koong, A.; Denko, N.C. Oxygen consumption can regulate the growth of tumors, a new perspective on the warburg effect. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Hardie, R.A.; van Dam, E.; Cowley, M.; Han, T.L.; Balaban, S.; Pajic, M.; Pinese, M.; Iconomou, M.; Shearer, R.F.; McKenna, J.; et al. Mitochondrial mutations and metabolic adaptation in pancreatic cancer. Cancer Metab. 2017, 5, 2. [Google Scholar] [CrossRef]
- Stoneking, M.; Delfin, F. The human genetic history of east Asia: Weaving a complex tapestry. Curr. Biol. 2010, 20, R188–R193. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of egfr regulates mitochondria dynamics and promotes metastasis in nsclc. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollu, L.R.; Ren, J.G.; Blessing, A.M.; Katreddy, R.R.; Gao, G.; Xu, L.; Wang, J.R.; Su, F.; Zhang, W.H. Involvement of de novo synthesized palmitate and mitochondrial egfr in egf induced mitochondrial fusion of cancer cells. Cell Cycle 2014, 13, 2415–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [Green Version]
- Agaronyan, K.; Morozov, Y.I.; Anikin, M.; Temiakov, D. Mitochondrial biology. Replication-transcription switch in human mitochondria. Science 2015, 347, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Fayzulin, R.Z.; Perez, M.; Kozhukhar, N.; Spadafora, D.; Wilson, G.L.; Alexeyev, M.F. A method for mutagenesis of mouse mtdna and a resource of mouse mtdna mutations for modeling human pathological conditions. Nucleic Acids Res. 2015, 43, e62. [Google Scholar] [CrossRef] [Green Version]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. Mtdna mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.Y.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct idh-mutant molecular profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Huang, S.F.; Liao, C.T.; Chen, I.H.; Wang, H.M.; Hsieh, L.L. Clinical significance in oral cavity squamous cell carcinoma of pathogenic somatic mitochondrial mutations. PLoS ONE 2013, 8, e65578. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Forced cytochrome b gene mutation expression induces mitochondrial proliferation and prevents apoptosis in human uroepithelial sv-huc-1 cells. Int. J. Cancer 2009, 125, 2829–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 18 April 2017).
- Patel, R.K.; Jain, M. Ngs qc toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491. [Google Scholar] [CrossRef]
- Navarro-Gomez, D.; Leipzig, J.; Shen, L.S.; Lott, M.; Stassen, A.P.M.; Wallace, D.C.; Wiggs, J.L.; Falk, M.J.; van Oven, M.; Gai, X.W. Phy-mer: A novel alignment-free and reference-independent mitochondrial haplogroup classifier. Bioinformatics 2015, 31, 1310–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | # Patients | % | Low Risk | High Risk | p-val | |
|---|---|---|---|---|---|---|
| Age | 1 | |||||
| =>65 | 39 | 63.9 | 22 | 17 | ||
| <65 | 22 | 36.1 | 13 | 9 | ||
| Sex | 0.1171 | |||||
| Male | 34 | 55.7 | 23 | 11 | ||
| Female | 27 | 44.3 | 12 | 15 | ||
| Smoking | 0.2763 | |||||
| Smokers | 19 | 31.2 | 13 | 6 | ||
| Never Smoked | 42 | 68.9 | 22 | 20 | ||
| Tumor Size | 0.5914 | |||||
| <=2 mm | 22 | 36.1 | 14 | 8 | ||
| >2 mm | 39 | 63.9 | 21 | 18 | ||
| EGFR mutation status | 0.6001 | |||||
| EGFR + | 36 | 59 | 22 | 14 | ||
| EGFR − | 25 | 41 | 13 | 12 | ||
| Relapse | 0.0003 | |||||
| Relapse | 21 | 34.4 | 5 | 16 | ||
| No Relapse | 40 | 65.6 | 30 | 10 | ||
| Survival | 0.5626 | |||||
| Survival | 45 | 73.8 | 27 | 18 | ||
| Death | 16 | 26.2 | 8 | 8 | ||
| Variable | adjHR | 95% CI | p-Value | |
|---|---|---|---|---|
| D-loop | ||||
| Age | 0.75 | 0.27 | 2.15 | 0.598 |
| Sex | 1.01 | 0.97 | 1.05 | 0.672 |
| EGFR mutation | 0.99 | 0.35 | 2.79 | 0.985 |
| Tumor Size | 3.74 | 0.99 | 14.07 | 0.051 |
| Smoking Status | 1.23 | 0.40 | 3.82 | 0.716 |
| D-loop mutations | 0.18 | 0.04 | 0.82 | 0.027 |
| Complex I | ||||
| Age | 0.99 | 0.95 | 1.03 | 0.573 |
| Sex | 0.89 | 0.31 | 2.57 | 0.827 |
| EGFR mutation | 0.78 | 0.28 | 2.16 | 0.633 |
| Tumor Size | 4.23 | 1.10 | 16.19 | 0.035 |
| Smoking Status | 0.99 | 0.32 | 3.04 | 0.983 |
| Complex-I mutations | 0.97 | 0.37 | 2.56 | 0.958 |
| Complex IV | ||||
| Age | 0.65 | 0.21 | 2.09 | 0.474 |
| Sex | 0.98 | 0.95 | 1.02 | 0.418 |
| EGFR mutation | 0.89 | 0.30 | 2.61 | 0.826 |
| Tumor Size | 3.99 | 1.13 | 14.14 | 0.032 |
| Smoking Status | 1.49 | 0.46 | 4.75 | 0.505 |
| Complex-IV mutations | 3.69 | 1.34 | 10.18 | 0.012 |
| Complex V | ||||
| Age | 0.62 | 0.21 | 1.81 | 0.379 |
| Sex | 0.97 | 0.93 | 1.01 | 0.141 |
| EGFR mutation | 0.58 | 0.19 | 1.73 | 0.324 |
| Tumor Size | 5.89 | 1.59 | 21.88 | 0.008 |
| Smoking Status | 1.05 | 0.32 | 3.37 | 0.941 |
| Complex-V mutations | 6.63 | 2.06 | 21.33 | 0.002 |
| Complex IV and Complex V | ||||
| Age | 0.60 | 0.18 | 1.95 | 0.391 |
| Sex | 0.98 | 0.94 | 1.02 | 0.325 |
| EGFR mutation | 0.91 | 0.31 | 2.72 | 0.869 |
| Tumor Size | 4.31 | 1.22 | 15.21 | 0.023 |
| Smoking Status | 2.07 | 0.63 | 6.86 | 0.233 |
| Complex IV and V mutations | 8.63 | 2.52 | 29.57 | 0.001 |
| Risk Score | ||||
| Age | 0.98 | 0.94 | 1.02 | 0.337 |
| Sex | 0.65 | 0.21 | 1.99 | 0.447 |
| EGFR mutation | 1.17 | 0.37 | 3.68 | 0.786 |
| Tumor Size | 4.03 | 1.11 | 14.66 | 0.034 |
| Smoking Status | 2.27 | 0.66 | 7.78 | 0.194 |
| High risk group | 9.10 | 2.93 | 28.32 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raghav, L.; Chang, Y.-H.; Hsu, Y.-C.; Li, Y.-C.; Chen, C.-Y.; Yang, T.-Y.; Chen, K.-C.; Hsu, K.-H.; Tseng, J.-S.; Chuang, C.-Y.; et al. Landscape of Mitochondria Genome and Clinical Outcomes in Stage 1 Lung Adenocarcinoma. Cancers 2020, 12, 755. https://doi.org/10.3390/cancers12030755
Raghav L, Chang Y-H, Hsu Y-C, Li Y-C, Chen C-Y, Yang T-Y, Chen K-C, Hsu K-H, Tseng J-S, Chuang C-Y, et al. Landscape of Mitochondria Genome and Clinical Outcomes in Stage 1 Lung Adenocarcinoma. Cancers. 2020; 12(3):755. https://doi.org/10.3390/cancers12030755
Chicago/Turabian StyleRaghav, Lovely, Ya-Hsuan Chang, Yi-Chiung Hsu, Yu-Cheng Li, Chih-Yi Chen, Tsung-Ying Yang, Kun-Chieh Chen, Kuo-Hsuan Hsu, Jeng-Sen Tseng, Cheng-Yen Chuang, and et al. 2020. "Landscape of Mitochondria Genome and Clinical Outcomes in Stage 1 Lung Adenocarcinoma" Cancers 12, no. 3: 755. https://doi.org/10.3390/cancers12030755