Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
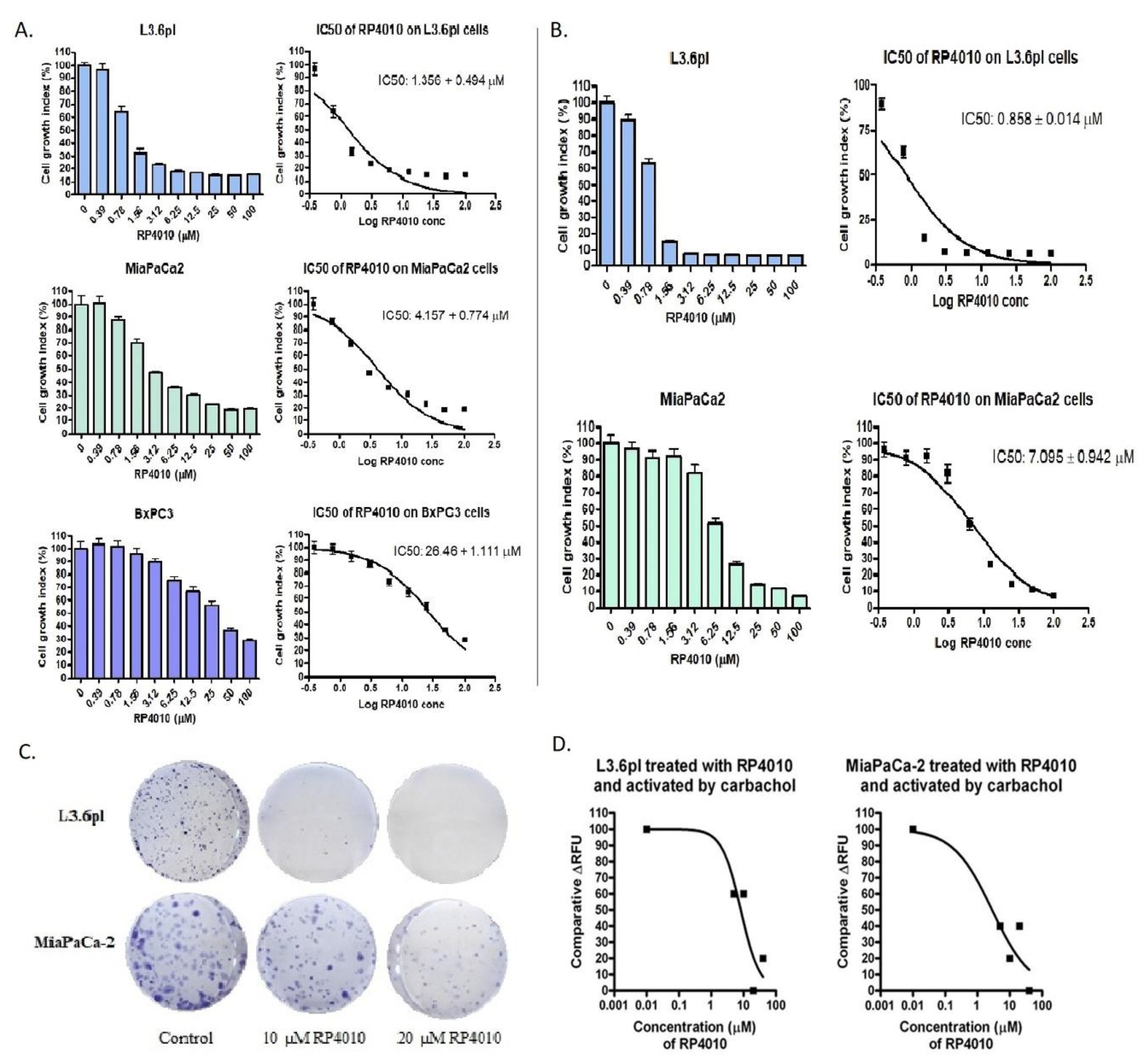
2.1. RP4010 Inhibited the Proliferation of Pancreatic Cancer Cells and Suppressed Pancreatic Cancer Cell Colony Formation
2.2. RP4010 Inhibited Carbachol-Induced Calcium Influx in Pancreatic Cancer Cells
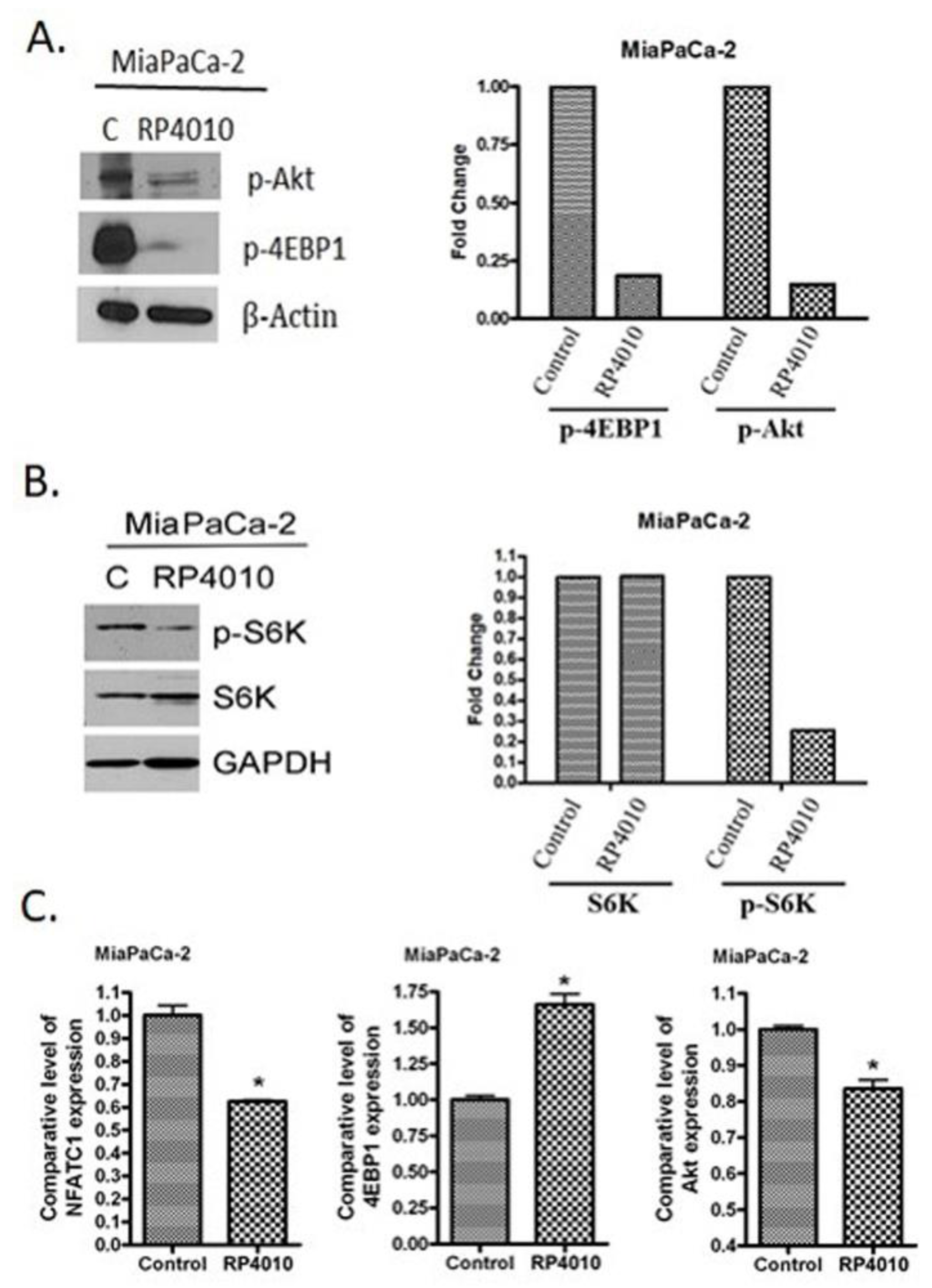
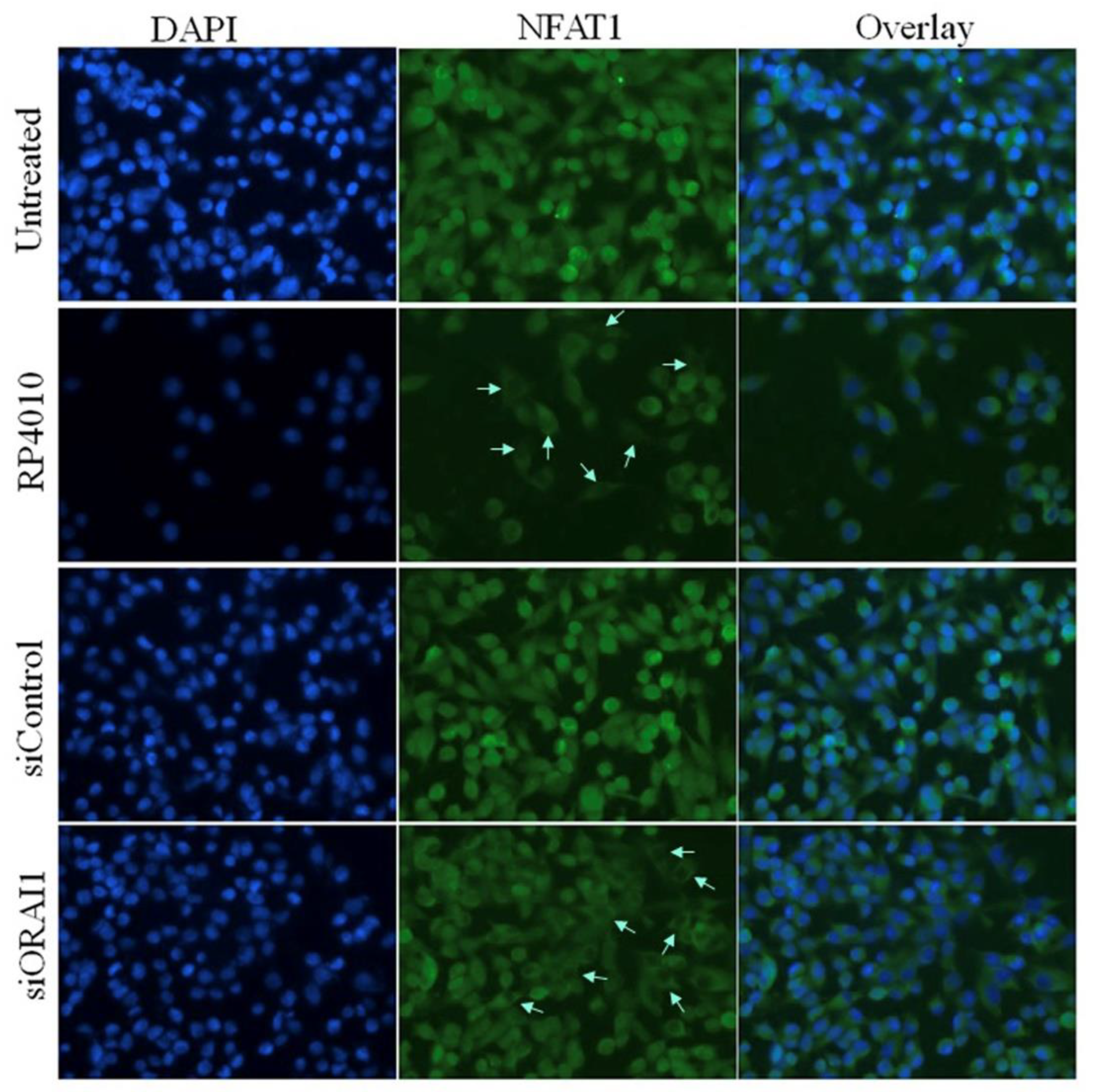
2.3. RP4010 Inhibited Calcium-Regulated Akt/mTOR and NFAT Signaling
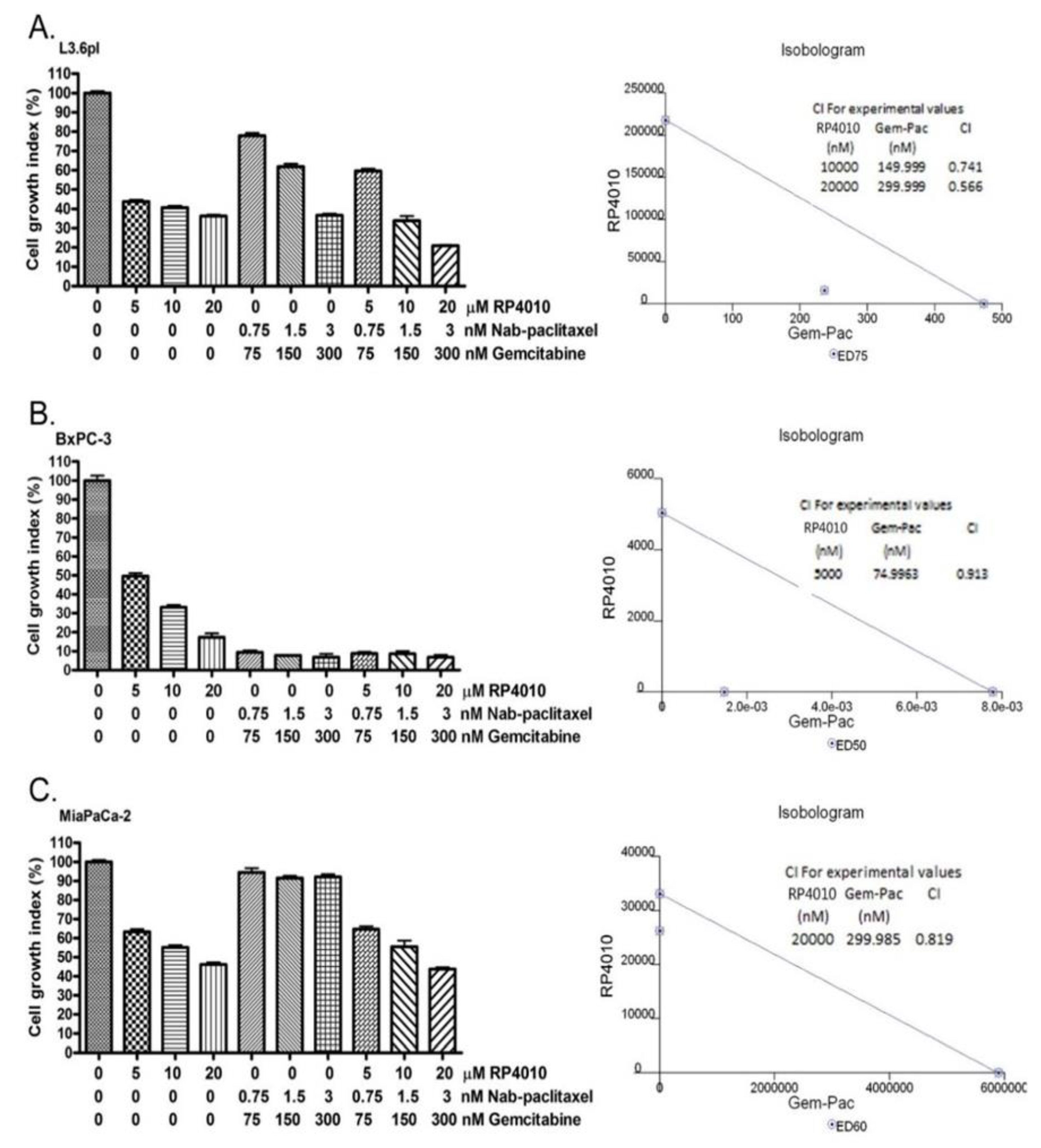
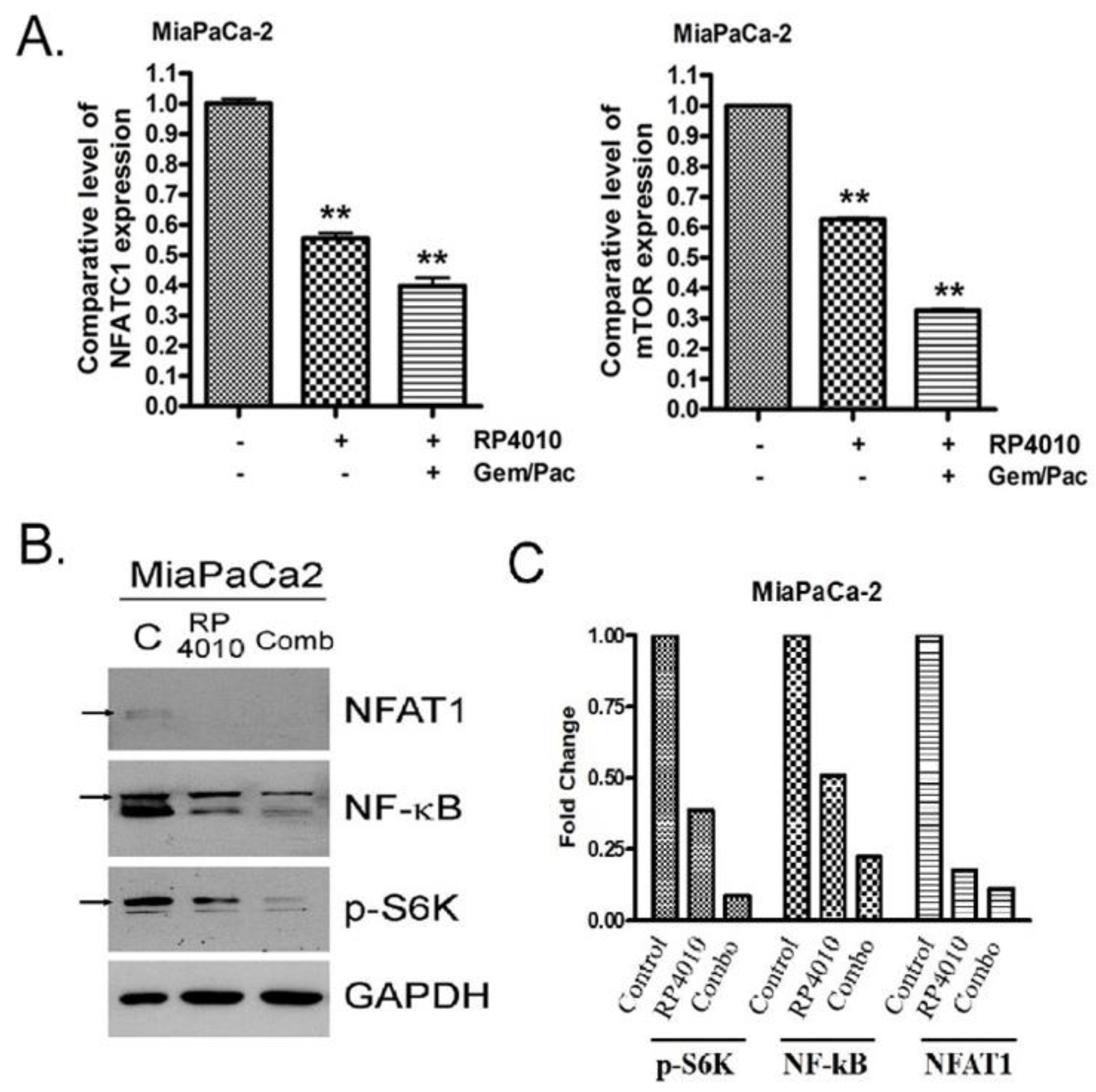
2.4. RP4010 and Gemcitabine/Nab-Paclitaxel Showed Synergistic Effects on the Inhibition of Cell Proliferation In Vitro with Downregulation of mTOR and NFAT/NF-κB Signaling
2.5. Anticancer Activity of RP4010 Is Potentiated by Gemcitabine/Nab-Paclitaxel in Patient-Derived Xenograft
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents and Antibodies
4.2. MTT Assay and Synergy Analysis
4.3. BrdU Cell Proliferation Assay
4.4. Small Interference RNA and Transfection
4.5. Cell Colony Formation Assay
4.6. Calcium Influx Assay
4.7. RNA Isolation and mRNA Real-Time RT-qPCR
4.8. Preparation of Total Protein Lysates and Western Blot Analysis
4.9. Immunofluorescence Analysis
4.10. Animal Studies
4.11. Immunostaining
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaal, C.; Padmanabhan, J.; Chellappan, S. The role of nAChR and calcium signaling in pancreatic cancer initiation and progression. Cancers 2015, 7, 1447–1471. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S.; Cahalan, M.D. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell. Regul. 1989, 1, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Malleter, M.; Tauzin, S.; Bessede, A.; Castellano, R.; Goubard, A.; Godey, F.; Levêque, J.; Jézéquel, P.; Campion, L.; Campone, M.; et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res. 2013, 73, 6711–6721. [Google Scholar] [CrossRef] [Green Version]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphaël, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Ishizuka, J.; Cooper, C.W.; Chung, D.H.; Tsuchiya, T.; Uchida, T.; Rajaraman, S.; Townsend, C.M., Jr.; Thompson, J.C. Inhibitory effect of calcium channel blockers on growth of pancreatic cancer cells. Pancreas 1994, 9, 193–202. [Google Scholar] [CrossRef]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Berry, C.T.; May, M.J.; Freedman, B.D. STIM- and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium 2018, 74, 131–143. [Google Scholar] [CrossRef]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Xie, J.; Pan, H.; Yao, J.; Zhou, Y.; Han, W. SOCE and cancer: Recent progress and new perspectives. Int. J. Cancer 2016, 138, 2067–2077. [Google Scholar] [CrossRef]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Future Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Franzius, D.; Hoth, M.; Penner, R. Non-specific effects of calcium entry antagonists in mast cells. Pflugers Arch. 1994, 428, 433–438. [Google Scholar] [CrossRef]
- Cui, C.; Chang, Y.; Zhang, X.; Choi, S.; Tran, H.; Penmetsa, K.V.; Viswanadha, S.; Fu, L.; Pan, Z. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 2018, 432, 169–179. [Google Scholar] [CrossRef]
- Anghileri, L.J.; Miller, E.S.; Robinette, J.; Prasad, K.N.; Lagerborg, V.A. Calcium metabolism in tumors. Its relationship with chromium complex accumulation. II. Calcium, magnesium and phosphorus in human and animal tumors. Oncology 1971, 25, 193–209. [Google Scholar] [CrossRef]
- Kraj, L.; Śliwczyński, A.; Krawczyk-Lipiec, J.; Woźniak, K.; Waszczuk-Gajda, A.; Rybski, S. Calcium channel blockers use and overall survival in pancreatic cancer patients receiving gemcitabine. J. Clin. Oncol. 2017, 35, e15756. [Google Scholar] [CrossRef]
- Wang, Z.; White, D.L.; Hoogeveen, R.; Chen, L.; Whitsel, E.A.; Richardson, P.A. Anti-Hypertensive Medication Use, Soluble Receptor for Glycation End Products and Risk of Pancreatic Cancer in the Women’s Health Initiative Study. J. Clin. Med. 2018, 7, 197. [Google Scholar] [CrossRef] [Green Version]
- Jäger, H.; Dreker, T.; Buck, A.; Giehl, K.; Gress, T.; Grissmer, S. Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 2004, 65, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhao, Y.; Schwarz, B.; Mysliwietz, J.; Hartig, R.; Camaj, P.; Bao, Q.; Jauch, K.W.; Guba, M.; Ellwart, J.W.; et al. Verapamil inhibits tumor progression of chemotherapy-resistant pancreatic cancer side population cells. Int. J. Oncol. 2016, 49, 99–110. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences | |
|---|---|---|
| NFATC1 | Forward | TGTCTGGGAGATGGAAGCGA |
| Reverse | CGGTTAGAAAGATGGCGTTACC | |
| Akt | Forward | TTGTGAAGGAGGGTTGGCTG |
| Reverse | CTCACGTTGGTCCACATCCT | |
| mTOR | Forward | TTCCGACCTTCTGCCTTCAC |
| Reverse | CCACAGAAAGTAGCCCCAGG | |
| 4EBP1 | Forward | CAAGGGATCTGCCCACCATT |
| Reverse | ACACGATGGCTGGTGCTTTA | |
| ORAI1 | Forward | GAGGTGATGAGCCTCAACGAG |
| Reverse | TAGTCGTGGTCAGCGTCCAG | |
| actin | Forward | GCACAGAGCCTCGCCTT |
| Reverse | TCATCATCCATGGTGAGCTG | |
| 18S | Forward | GCAATTATTCCCCATGAACG |
| Reverse | GGCCTCACTAAACCATCCAA | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, H.Y.; Mpilla, G.B.; Sexton, R.; Viswanadha, S.; Penmetsa, K.V.; Aboukameel, A.; Diab, M.; Kamgar, M.; Al-Hallak, M.N.; Szlaczky, M.; et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers 2020, 12, 750. https://doi.org/10.3390/cancers12030750
Khan HY, Mpilla GB, Sexton R, Viswanadha S, Penmetsa KV, Aboukameel A, Diab M, Kamgar M, Al-Hallak MN, Szlaczky M, et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers. 2020; 12(3):750. https://doi.org/10.3390/cancers12030750
Chicago/Turabian StyleKhan, Husain Yar, Gabriel B. Mpilla, Rachel Sexton, Srikant Viswanadha, Kumar V. Penmetsa, Amro Aboukameel, Maria Diab, Mandana Kamgar, Mohammed Najeeb Al-Hallak, Mark Szlaczky, and et al. 2020. "Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth" Cancers 12, no. 3: 750. https://doi.org/10.3390/cancers12030750