Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety

 , , and
, , and
Abstract
:1. Introduction
2. Results
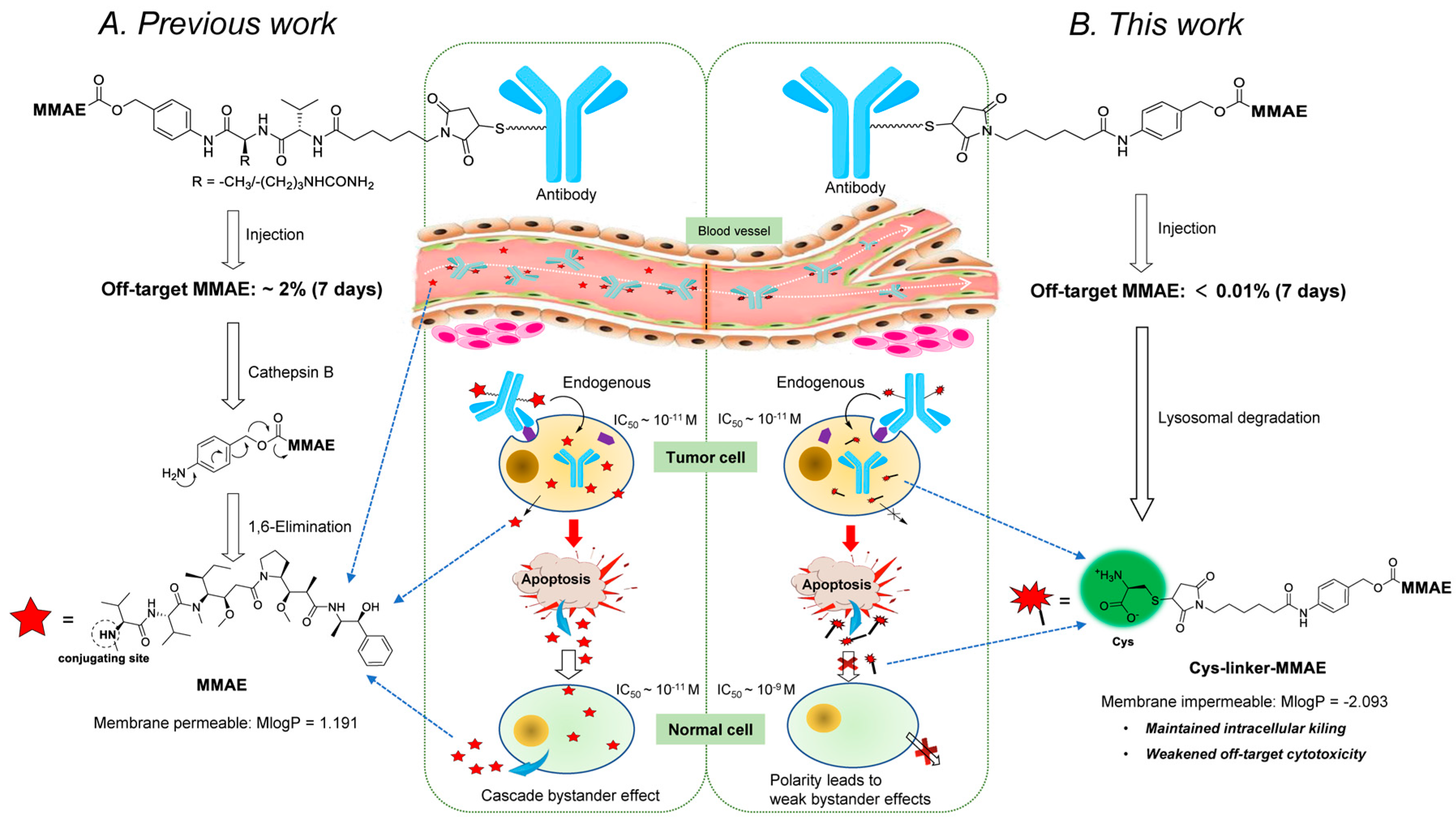
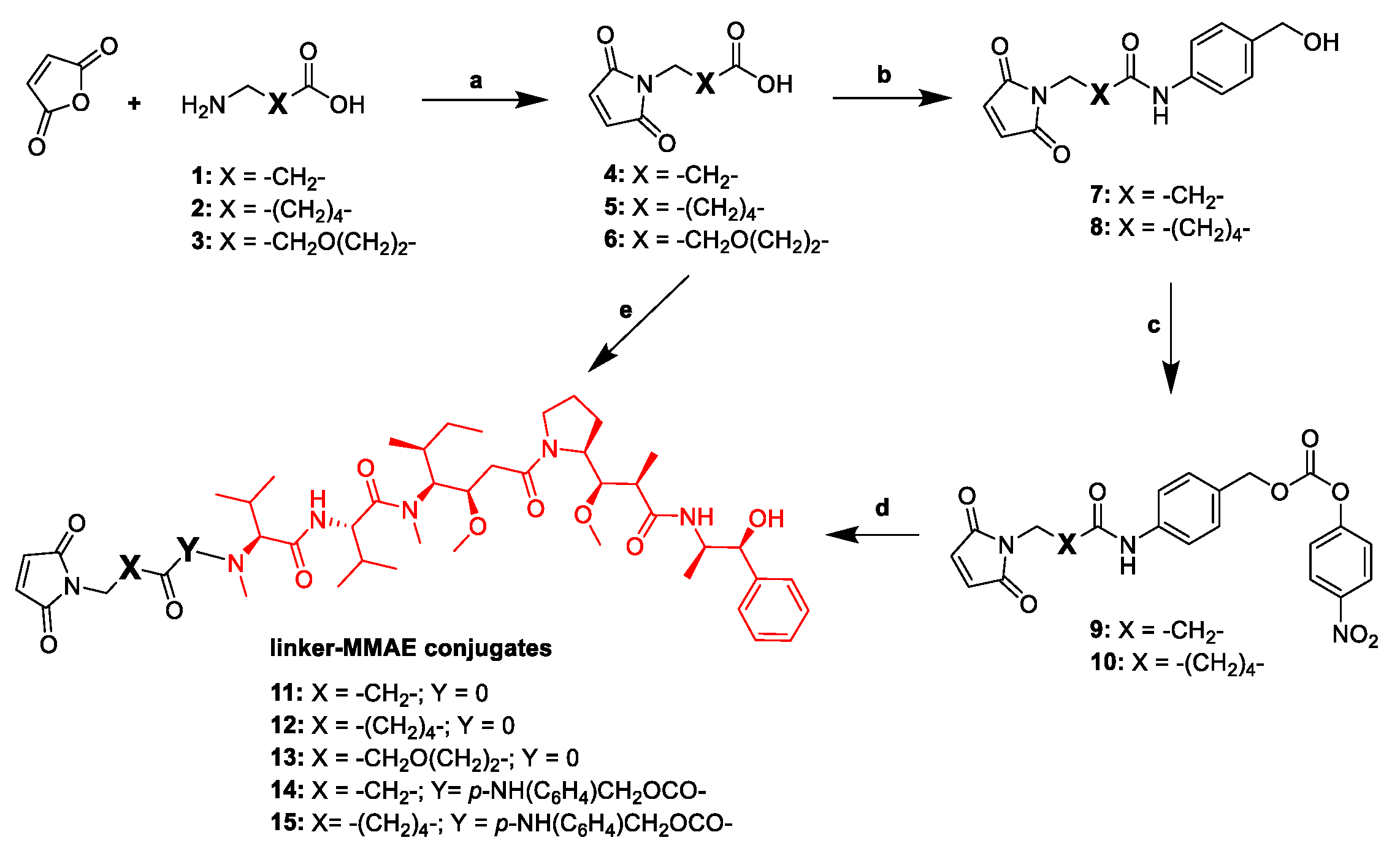
2.1. Design and Synthesis of the Cys-Linker-MMAE-Based ADC Payloads
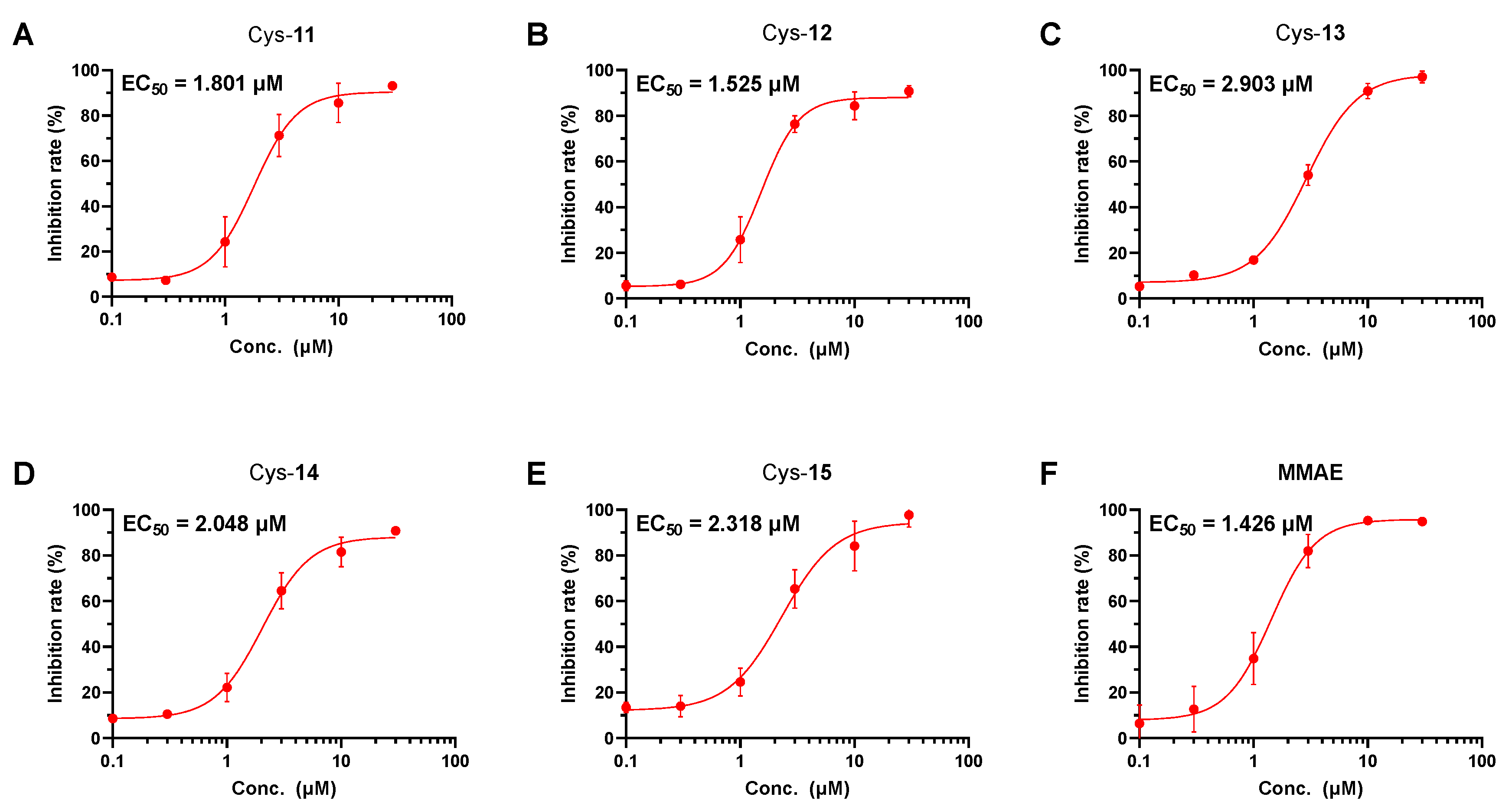
2.2. Tubulin Polymerization Inhibition Study
2.3. Evaluation of the Antibody-Drug Conjugates (ADCs) Preparation
2.4. Stability Assays of the Conjugate in Plasma
2.5. Drug Release Study at the Cellular Level
2.6. Flow Cytometry Analysis
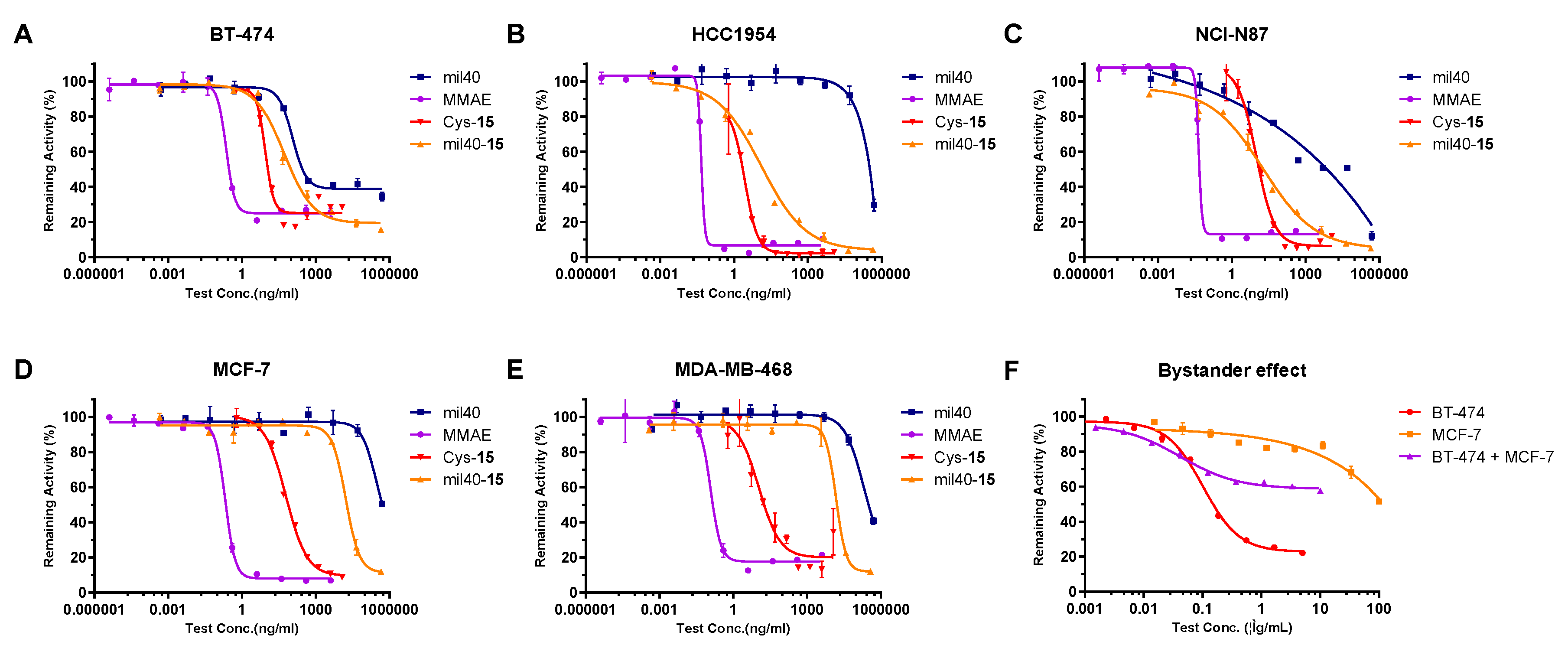
2.7. In Vitro Potency Assay of the ADC, Antibody, MMAE, and Cys-Linker-MMAE Conjugate
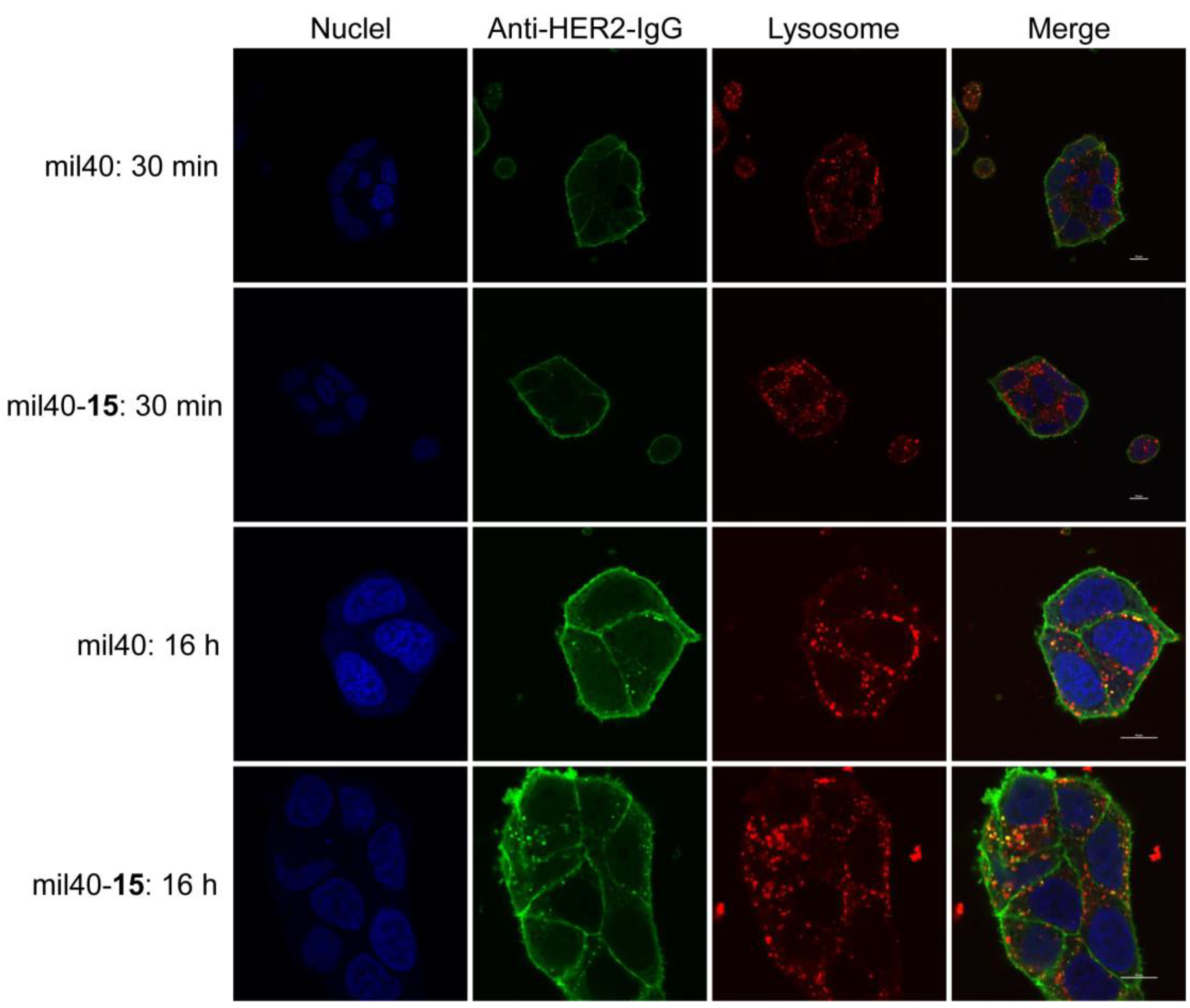
2.8. Trafficking Assay by Fluorescence Microscopy
2.9. In Vivo Potency in Xenografted Nude Mice
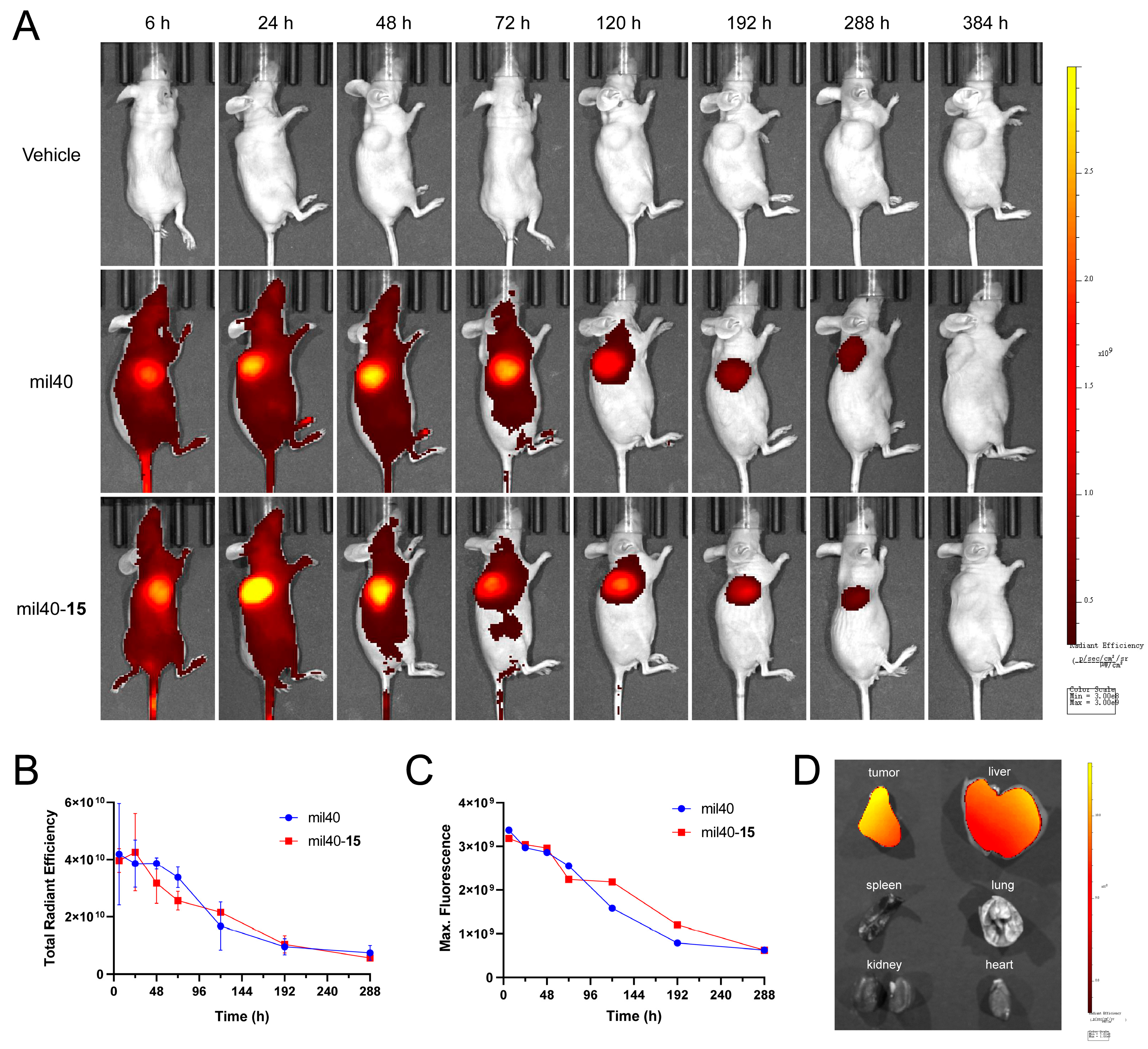
2.10. In Vivo Imaging of Fluorescein-Labeled ADC
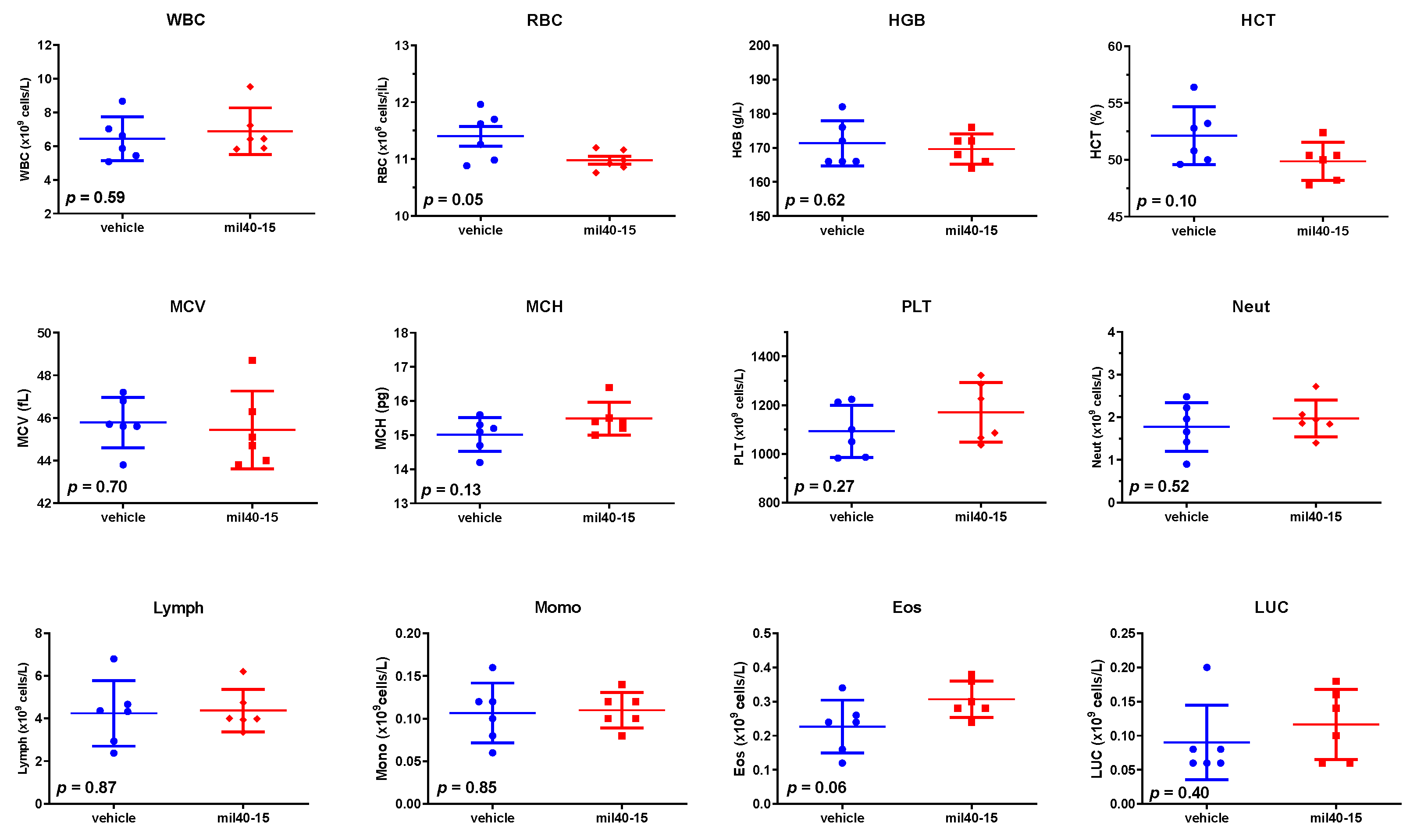
2.11. Hematological Analysis
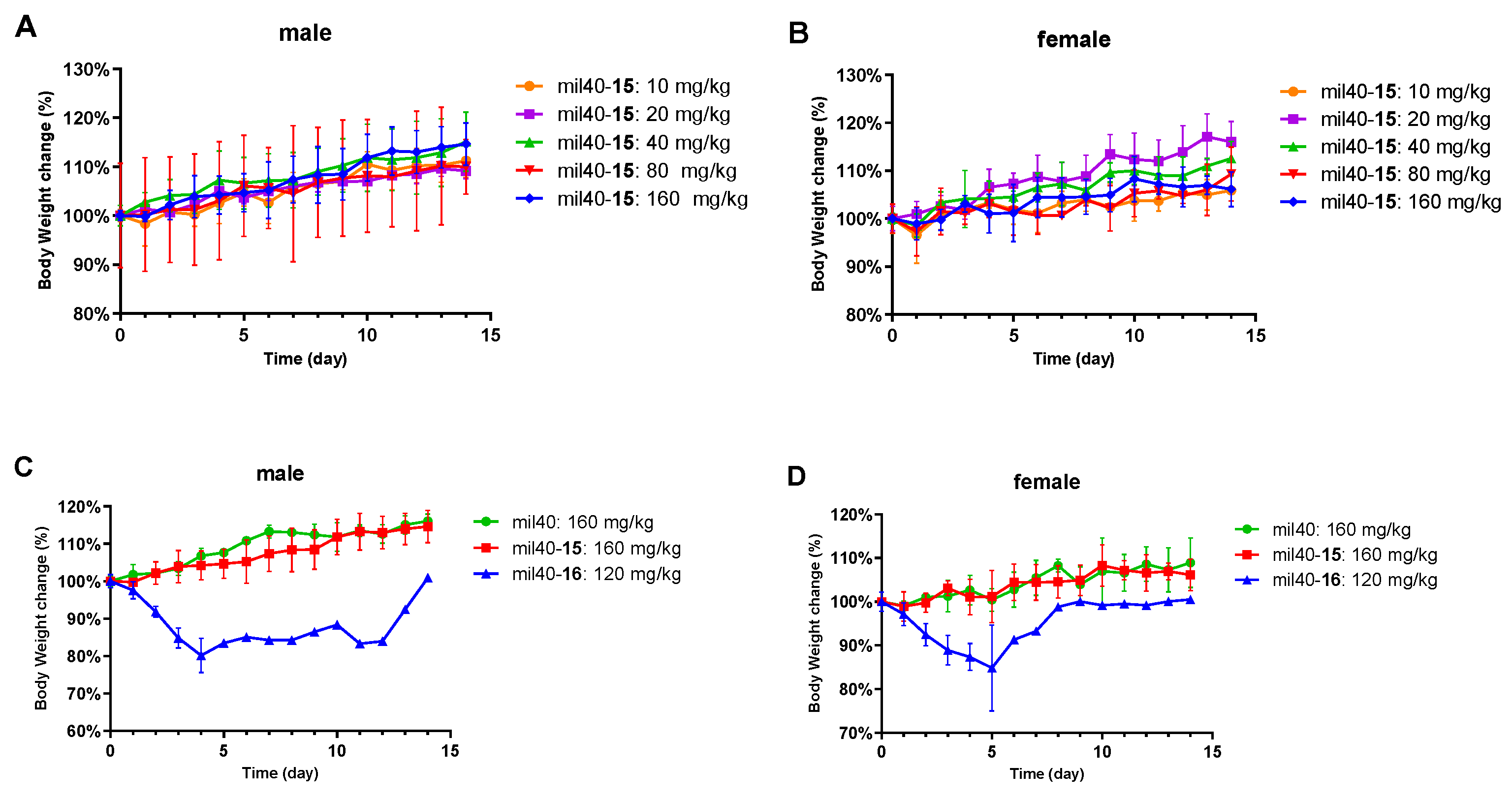
2.12. In Vivo Tolerability Study
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibody
4.2. Synthesis of the Linker-MMAE Conjugates
4.2.1. Synthesis of 3-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Propanoic Acid (4)
4.2.2. Synthesis of 6-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Hexanoic Acid (5)
4.2.3. Synthesis of 3-(2-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Ethoxy)Propanoic Acid (6)
4.2.4. Synthesis of 3-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)-N-(4-(Hydroxymethyl)Phenyl)Propanamide (7)
4.2.5. Synthesis of 6-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)-N-(4-(Hydroxymethyl)Phenyl)Hexanamide (8)
4.2.6. Synthesis of 4-(3-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Propanamido)Benzyl (4-Nitrophenyl) Carbonate (9)
4.2.7. Synthesis of 4-(6-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Hexanamido)Benzyl (4-Nitrophenyl) Carbonate (10)
4.2.8. Synthesis of (S)-2-((S)-2-(3-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)-N-Methylpropanamido)-3-Methylbutanamido)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-Hydroxy-1-Phenylpropan-2-yl)amino)-1-Methoxy-2-Methyl-3-Oxopropyl)Pyrrolidin-1-yl)-3-Methoxy-5-Methyl-1-Oxoheptan-4-yl)-N,3-Dimethylbutanamide (11)
4.2.9. Synthesis of 6-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)-N-((S)-1-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-Hydroxy-1-Phenylpropan-2-yl)Amino)-1-Methoxy-2-Methyl-3-Oxopropyl)Pyrrolidin-1-yl)-3-Methoxy-5-Methyl-1-Oxoheptan-4-yl)(Methyl)Amino)-3-Methyl-1-Oxobutan-2-yl)Amino)-3-Methyl-1-Oxobutan-2-yl)-N-Methylhexanamide (12)
4.2.10. Synthesis of 4-(3-(2-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Ethoxy)Propanamido)Benzyl ((S)-1-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-Hydroxy-1-Phenylpropan-2-yl)Amino)-1-Methoxy-2-Methyl-3-Oxopropyl)Pyrrolidin-1-yl)-3-Methoxy-5-Methyl-1-Oxoheptan-4-yl)(Methyl)Amino)-3-Methyl-1-Oxobutan-2-yl)Amino)-3-Methyl-1-Oxobutan-2-yl)(Methyl)Carbamate (13)
4.2.11. Synthesis of 4-(3-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Propanamido)Benzyl ((S)-1-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-Hydroxy-1-Phenylpropan-2-yl)Amino)-1-Methoxy-2-Methyl-3-Oxopropyl)Pyrrolidin-1-yl)-3-Methoxy-5-Methyl-1-Oxoheptan-4-yl)(Methyl)Amino)-3-Methyl-1-Oxobutan-2-yl)Amino)-3-Methyl-1-Oxobutan-2-yl)(Methyl)Carbamate (14)
4.2.12. Synthesis of 4-(6-(2,5-Dioxo-2,5-Dihydro-1H-Pyrrol-1-yl)Hexanamido)Benzyl ((S)-1-(((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-Hydroxy-1-Phenylpropan-2-yl)Amino)-1-Methoxy-2-Methyl-3-Oxopropyl)Pyrrolidin-1-yl)-3-Methoxy-5-Methyl-1-Oxoheptan-4-yl)(Methyl)Amino)-3-Methyl-1-Oxobutan-2-yl)Amino)-3-Methyl-1-Oxobutan-2-yl)(Methyl)Carbamate (15)
4.3. Microtubule Polymerization Assay
4.4. Preparation of Reagents and Antibody-Drug Conjugates
4.5. Characterization of the ADC
4.6. Stability Assays of the ADC in Plasma
4.7. Cellular Drug Release Process
4.8. Evaluation of the ADC for Tumor Cell Killing In Vitro
4.9. Confocal Analysis for Intracellular Localization
4.10. Cell Cycle Arrest and Apoptosis Analysis
4.11. In Vivo Antitumor Activity in Human Gastric Xenograft Tumors
4.12. In Vivo Fluorescence Imaging Experiment
4.13. Hematological Analysis during Treatment
4.14. In Vivo Tolerability Experiments
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Maaheimo, H.; Ekholm, F.S. New insight on the structural features of the cytotoxic auristatins mmae and mmaf revealed by combined nmr spectroscopy and quantum chemical modelling. Sci. Rep. 2017, 7, e15920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerr, C.; Friess, W. Antibody-drug conjugates- stability and formulation. Eur. J. Pharm. Biopharm. 2019, 139, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R. Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.S.; Tice, D.A.; Soria, J.C. Antibody-drug conjugates: Future directions in clinical and translational strategies to improve the therapeutic index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, S.; Xiao, D.; Xie, F.; Li, W.; Zhong, W.; Zhou, X. Novel silyl ether-based acid-cleavable antibody-mmae conjugates with appropriate stability and efficacy. Cancers 2019, 11, 957. [Google Scholar] [CrossRef] [Green Version]
- Breij, E.C.; de Goeij, B.E.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An antibody-drug conjugate that targets tissue factor exhibits potent therapeutic activity against a broad range of solid tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Walles, M.; Connor, A.; Hainzl, D. Adme and safety aspects of non-cleavable linkers in drug discovery and development. Curr. Top Med. Chem. 2017, 17, 3463–3475. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (t-dm1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug. Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Mort, J.S. Cathepsin b. In Handbook of Proteolytic Enzymes; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1784–1791. [Google Scholar]
- Bornstein, G.G. Antibody drug conjugates: Preclinical considerations. AAPS J. 2015, 17, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [Green Version]
- Cini, E.; Faltoni, V.; Petricci, E.; Taddei, M.; Salvini, L.; Giannini, G.; Vesci, L.; Milazzo, F.M.; Anastasi, A.M.; Battistuzzi, G.; et al. Antibody drug conjugates (adcs) charged with hdac inhibitor for targeted epigenetic modulation. Chem. Sci. 2018, 9, 6490–6496. [Google Scholar] [CrossRef] [Green Version]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat. Commun. 2018, 9, e2512. [Google Scholar] [CrossRef]
- Alley, S.C.; Zhang, X.; Okeley, N.M.; Anderson, M.; Law, C.L.; Senter, P.D.; Benjamin, D.R. The pharmacologic basis for antibody-auristatin conjugate activity. J. Pharmacol. Exp. Ther. 2009, 330, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting her2-positive breast cancer with trastuzumab-dm1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin f through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, S.; Zhong, W.; Zhou, X.; Li, S. Development and properties of valine-alanine based antibody-drug conjugates with monomethyl auristatin e as the potent payload. Int. J. Mol. Sci. 2017, 18, 1860. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Liu, Y.N.; Wang, J.J.; Ji, Y.T.; Zhao, G.D.; Tang, L.Q.; Zhang, C.M.; Guo, X.L.; Liu, Z.P. Design, synthesis, and biological evaluation of 1-methyl-1,4-dihydroindeno[1,2-c]pyrazole analogues as potential anticancer agents targeting tubulin colchicine binding site. J. Med. Chem. 2016, 59, 5341–5355. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. Cac10-vcmmae, an anti-cd30-monomethyl auristatin e conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Petit, E.; Bosch, L.; Costa, A.M.; Vilarrasa, J. (z)-oxopropene-1,3-diyl, a linker for the conjugation of the thiol group of cysteine with amino-derivatized drugs. J. Org. Chem. 2019, 84, 11170–11176. [Google Scholar] [CrossRef]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blattler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [Green Version]
- Nunes, J.P.M.; Vassileva, V.; Robinson, E.; Morais, M.; Smith, M.E.B.; Pedley, R.B.; Caddick, S.; Baker, J.R.; Chudasama, V. Use of a next generation maleimide in combination with thiomab™ antibody technology delivers a highly stable, potent and near homogeneous thiomab™ antibody-drug conjugate (tdc). RSC Adv. 2017, 7, 24828–24832. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szot, C.; Saha, S.; Zhang, X.M.; Zhu, Z.; Hilton, M.B.; Morris, K.; Seaman, S.; Dunleavey, J.M.; Hsu, K.S.; Yu, G.J.; et al. Tumor stroma-targeted antibody-drug conjugate triggers localized anticancer drug release. J. Clin. Investig. 2018, 128, 2927–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Corso, A.; Cazzamalli, S.; Gebleux, R.; Mattarella, M.; Neri, D. Protease-cleavable linkers modulate the anticancer activity of noninternalizing antibody-drug conjugates. Bioconjug. Chem. 2017, 28, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, B.A.; Barnscher, S.D.; Snyder, J.T.; An, Z.; Dodd, J.M.; Dugal-Tessier, J. Investigation of hydrophilic auristatin derivatives for use in antibody drug conjugates. Bioconjug. Chem. 2017, 28, 371–381. [Google Scholar] [CrossRef]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of sgn-35, a potent anti-cd30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (adcs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linker-MMAE |  | |
|---|---|---|
| X | Y | |
| 11 | -CH2- | - |
| 12 | -(CH2)4- | - |
| 13 | -CH2O(CH2)2- | - |
| 14 | -CH2- |  |
| 15 | -(CH2)4- |  |
| 16 | -(CH2)4- |  |
| Cell Lines | HER2 Status | mil40-15 | mil40 | MMAE | Cys-15 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | Max. Inhibition | IC50 (nM) | Max. Inhibition | IC50(nM) | Max. Inhibition | IC50 (nM) | Max. Inhibition | ||
| BT-474 | HER2+++ | 0.35 | 84.49% | 0.78 | 65.45% | 0.32 | 79.09% | 6.88 | 82.62% |
| HCC1954 | HER2+++ | 0.09 | 96.29% | 1753.53 | 70.36% | 0.07 | 97.54% | 2.31 | 98.86% |
| NCI-N87 | HER2+++ | 0.12 | 95.01% | 0.19 | 87.90% | 0.07 | 89.37% | 8.67 | 94.86% |
| MCF-7 | HER2− | 110.54 | 88.06% | >3438.25 | 49.43% | 0.29 | 93.14% | 51.23 | 91.13% |
| MDA-MB-468 | HER2− | 97.62 | 87.42% | 1388.43 | 59.13% | 0.17 | 87.37% | 11.11 | 85.75% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, L.; Fan, S.; Xiao, D.; Xie, F.; Li, W.; Zhong, W.; Zhou, X. Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety. Cancers 2020, 12, 744. https://doi.org/10.3390/cancers12030744
Wang Y, Liu L, Fan S, Xiao D, Xie F, Li W, Zhong W, Zhou X. Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety. Cancers. 2020; 12(3):744. https://doi.org/10.3390/cancers12030744
Chicago/Turabian StyleWang, Yanming, Lianqi Liu, Shiyong Fan, Dian Xiao, Fei Xie, Wei Li, Wu Zhong, and Xinbo Zhou. 2020. "Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety" Cancers 12, no. 3: 744. https://doi.org/10.3390/cancers12030744