The Capacity of High-Grade Serous Ovarian Cancer Cells to Form Multicellular Structures Spontaneously along Disease Progression Correlates with Their Orthotopic Tumorigenicity in Immunosuppressed Mice



Abstract
:
1. Introduction
2. Results
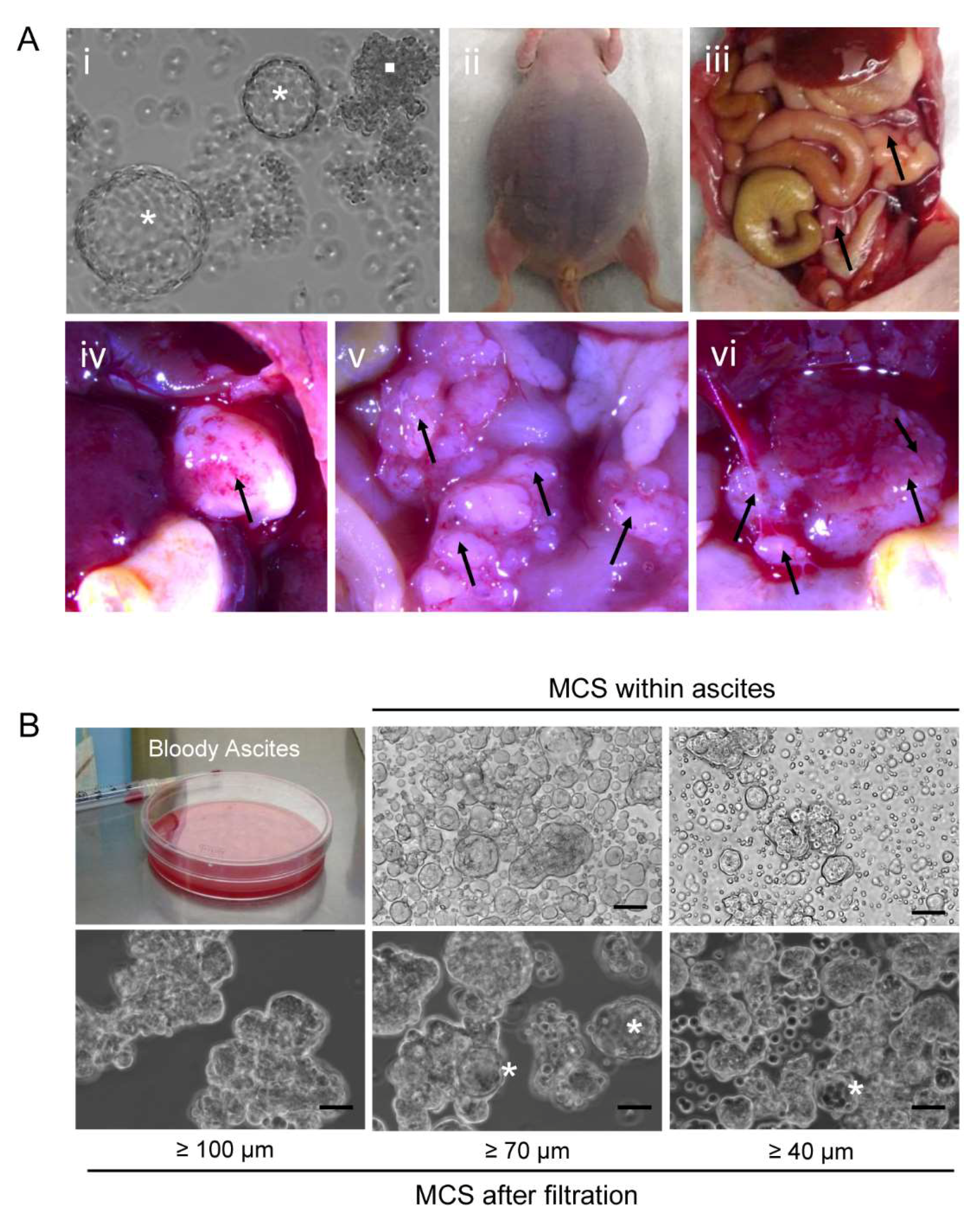
2.1. Serous Ovarian Cancer Cells of Advanced Disease Self-Assemble into Multicellular Structures in Suspension Despite Being Cultured in Anchorage-Prone Conditions
2.2. Expression of Biomarkers in High-Grade Serous Ovarian Cancer Cells along Disease Progression
2.3. Both Flat Adherent Cells and Cells Within Multicellular Structures Have Proliferative Capacity and Express E-Cadherin
2.4. Free-Floating Multicellular Structures Are Viable Entities
2.5. The Free-Floating Multicellular Structures Generated Spontaneously from a Culture of PEO6 Cells Display a Coral-Like or Irregular Phenotype Together with More Organized Spheroidal Arrangement
2.6. Non-Adherent Ovarian Cancer Multicellular Structures of Spheroidal Nature Have the Capacity to Perpetuate the Entire Phenotype when Moved to a New Culture Environment
2.7. PEO1, PEO4, and PEO6 Cells Have Differential Tumorigenicity in Vivo
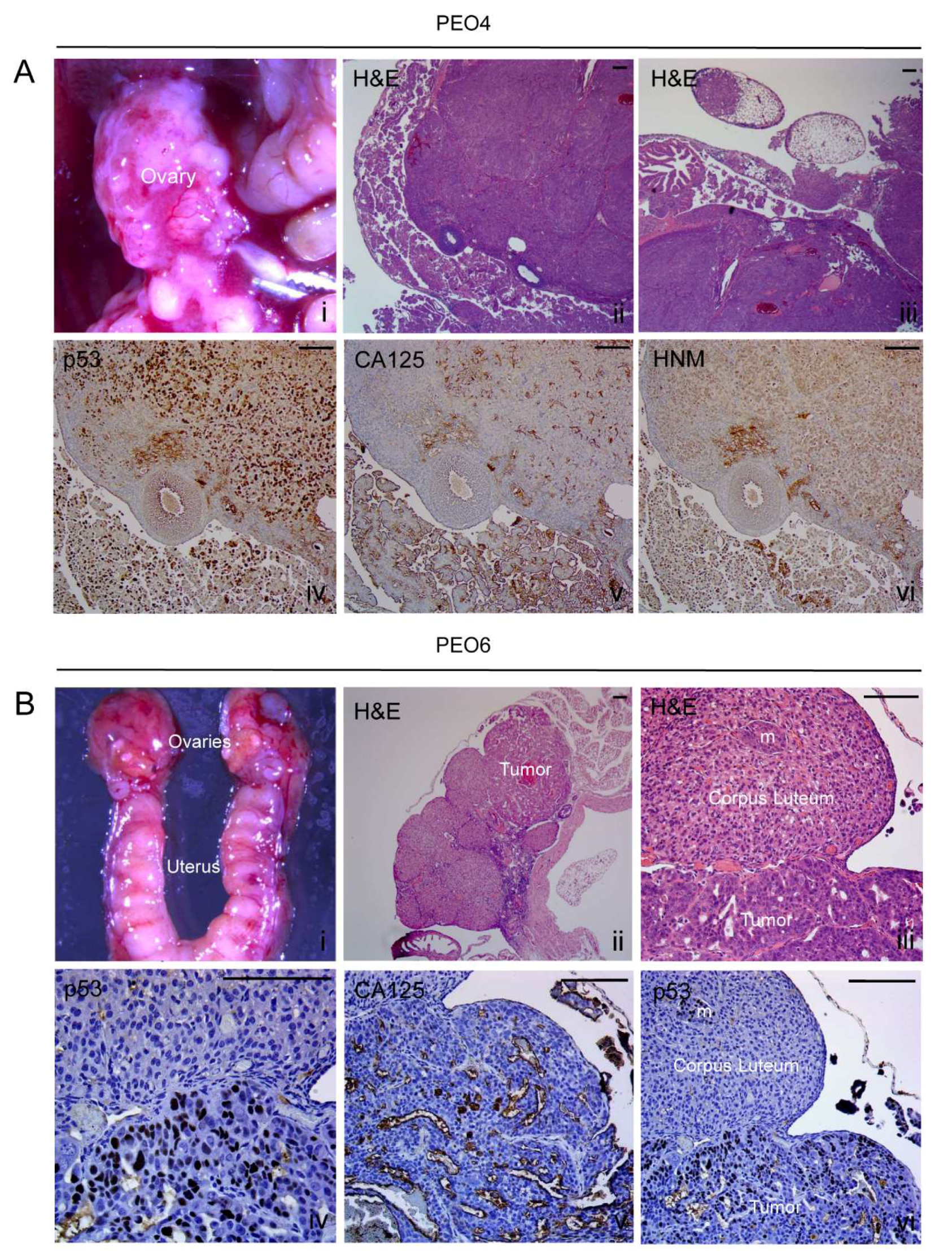
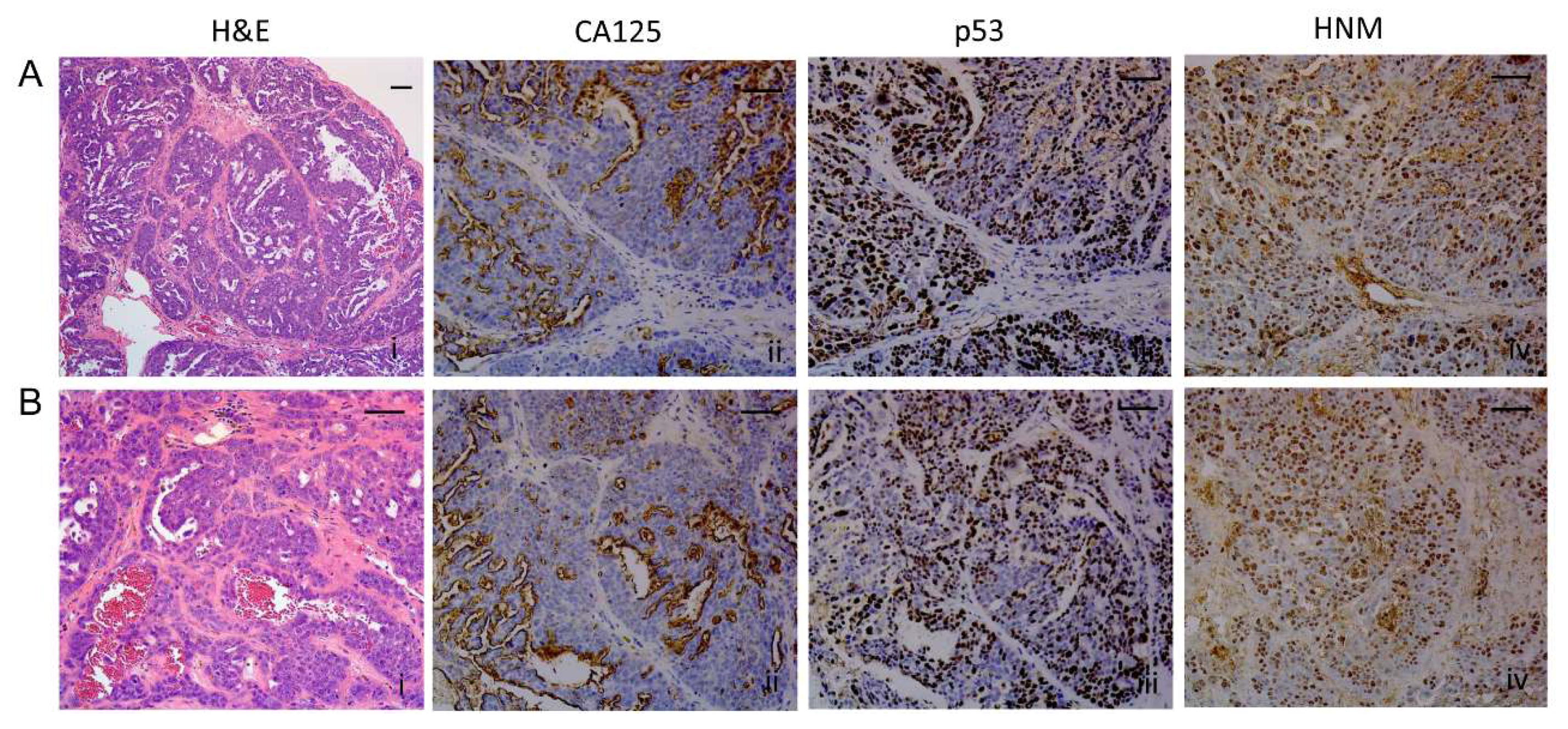
2.8. Histopathological Assessment of Intraperitoneal Tumors Derived from PEO4 or PEO6 Cells
2.8.1. Diaphragm
2.8.2. Omentum
2.8.3. Ovary
2.8.4. Liver
2.8.5. Peritoneal Wall
2.9. Orthometastatic Injection of PEO6 Multicellular Structures Is Sufficient to Trigger the Development of Bloody Ascites and Peritoneal Disease
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Line Authentication
4.3. Phase Contrast Microscopy
4.4. Cytospin Preparations
4.5. Fluorescence Microscopy
4.6. Immunocytochemistry and Immunohistochemistry
4.7. Studies in Immunosuppressed Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Cell Line Validation via STR Assessment

References
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Naora, H.; Montell, D.J. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Coleman, R.L.; Monk, B.J.; Sood, A.K.; Herzog, T.J. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013, 10, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Puls, L.E.; Duniho, T.; Hunter, J.E.; Kryscio, R.; Blackhurst, D.; Gallion, H. The prognostic implication of ascites in advanced-stage ovarian cancer. Gynecol. Oncol. 1996, 61, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Risberg, B.; Reich, R.; Berner, A. Effusion cytology in ovarian cancer: New molecular methods as aids to diagnosis and prognosis. Clin. Lab. Med. 2003, 23, 729–754. [Google Scholar] [CrossRef]
- Zuna, R.E.; Behrens, A. Peritoneal washing cytology in gynecologic cancers: Long-term follow-up of 355 patients. J. Natl. Cancer Inst. 1996, 88, 980–987. [Google Scholar] [CrossRef]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Krugmann, J.; Schwarz, C.L.; Melcher, B.; Sterlacci, W.; Ozalinskaite, A.; Lermann, J.; Agaimy, A.; Vieth, M. Malignant ascites occurs most often in patients with high-grade serous papillary ovarian cancer at initial diagnosis: A retrospective analysis of 191 women treated at Bayreuth Hospital, 2006–2015. Arch. Gynecol. Obstet. 2019, 299, 515–523. [Google Scholar] [CrossRef]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [Green Version]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar] [PubMed]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Emori, M.M.; Papp, E.; MacDuffie, E.; Konecny, G.E.; Velculescu, V.E.; Drapkin, R. Beyond genomics: Critical evaluation of cell line utility for ovarian cancer research. Gynecol. Oncol. 2015, 139, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y.; et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Takeda, T.; Banno, K.; Okawa, R.; Yanokura, M.; Iijima, M.; Irie-Kunitomi, H.; Nakamura, K.; Iida, M.; Adachi, M.; Umene, K.; et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol. Rep. 2016, 35, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [Green Version]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Bast, R.C., Jr.; Spriggs, D.R. More than a biomarker: CA125 may contribute to ovarian cancer pathogenesis. Gynecol. Oncol. 2011, 121, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.J.; Porter, C.; Gamarra, M.; Piver, M.S.; Johnson, E.A. Isolation and morphologic characterization of human ovarian carcinoma cell clusters present in effusions. Exp. Cell Biol. 1987, 55, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Pease, J.C.; Brewer, M.; Tirnauer, J.S. Spontaneous spheroid budding from monolayers: A potential contribution to ovarian cancer dissemination. Biol. Open 2012, 1, 622–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Berner, A.; Nesland, J.M.; Risberg, B.; Berner, H.S.; Trope, C.G.; Kristensen, G.B.; Bryne, M.; Ann Florenes, V. E-cadherin and alpha-, beta-, and gamma-catenin protein expression is up-regulated in ovarian carcinoma cells in serous effusions. J. Pathol. 2000, 192, 460–469. [Google Scholar] [CrossRef]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Davidowitz, R.A.; Selfors, L.M.; Iwanicki, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef] [Green Version]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef]
- Burleson, K.M.; Boente, M.P.; Pambuccian, S.E.; Skubitz, A.P. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J. Transl. Med. 2006, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Goyeneche, A.A.; Telleria, C.M. Ovarian Cancer Research in the Post Genomic Era—Challenges and Opportunities. In Gynecologic Cancers—Basic Sciences, Clinical and Therapeutic Perspectives; Farghaly, S.A., Ed.; InTechOpen: Rijeka, Croatia, 2016; pp. 149–163. [Google Scholar]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bapat, S.A. Human ovarian cancer stem cells. Reproduction 2010, 140, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [Green Version]
- Maitland, N.J.; Collins, A.T. Prostate cancer stem cells: A new target for therapy. J. Clin. Oncol. 2008, 26, 2862–2870. [Google Scholar] [CrossRef]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xie, J.; Guo, J.; Manning, H.C.; Gore, J.C.; Guo, N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 2012, 28, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T.; Huang, Z.; Bentley, R.C.; Mori, S.; Fujii, S.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Wintzell, M.; Hjerpe, E.; Avall Lundqvist, E.; Shoshan, M. Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 2012, 12, 359. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2012, 130, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Edwards, N.S.; Yates, J.D. Spheroids and cell survival. Crit. Rev. Oncol. Hematol. 2000, 36, 61–74. [Google Scholar] [CrossRef]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef]
- Shield, K.; Riley, C.; Quinn, M.A.; Rice, G.E.; Ackland, M.L.; Ahmed, N. Alpha2beta1 integrin affects metastatic potential of ovarian carcinoma spheroids by supporting disaggregation and proteolysis. J. Carcinog. 2007, 6, 11. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [Green Version]
- Davidson, B.; Reich, R.; Trope, C.G.; Wang, T.L.; Shih Ie, M. New determinates of disease progression and outcome in metastatic ovarian carcinoma. Histol. Histopathol. 2010, 25, 1591–1609. [Google Scholar]
- Zietarska, M.; Maugard, C.M.; Filali-Mouhim, A.; Alam-Fahmy, M.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol. Carcinog. 2007, 46, 872–885. [Google Scholar] [CrossRef]
- Correa, R.J.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis 2012, 33, 49–58. [Google Scholar] [CrossRef]
- Hamilton, T.C.; Young, R.C.; Louie, K.G.; Behrens, B.C.; McKoy, W.M.; Grotzinger, K.R.; Ozols, R.F. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice. Cancer Res. 1984, 44, 5286–5290. [Google Scholar] [PubMed]
- Ward, B.G.; Wallace, K.; Shepherd, J.H.; Balkwill, F.R. Intraperitoneal xenografts of human epithelial ovarian cancer in nude mice. Cancer Res. 1987, 47, 2662–2667. [Google Scholar] [PubMed]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Jcken, W.F.J.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Weroha, S.J.; Becker, M.A.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; AlHilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J., Jr.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, X.; Shi, G.; Xie, X.; Liu, H.; Zhang, X.; Lai, Y.; Zuo, Y.; Chen, Z.; Liu, S.; et al. Establishment of a new representative model of human ovarian cancer in mice. J. Ovarian Res. 2013, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Coffman, L.G.; Pearson, A.T.; Frisbie, L.G.; Freeman, Z.; Christie, E.; Bowtell, D.D.; Buckanovich, R.J. Ovarian Carcinoma-Associated Mesenchymal Stem Cells Arise from Tissue-Specific Normal Stroma. Stem Cells 2019, 37, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Batra, S.K. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer Res. 2015, 75, 4669–4674. [Google Scholar] [CrossRef] [Green Version]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyeneche, A.A.; Harmon, J.M.; Telleria, C.M. Cell death induced by serum deprivation in luteal cells involves the intrinsic pathway of apoptosis. Reproduction 2006, 131, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Ascites | Peritoneal Disease | Time to Euthanasia (Months) | Type of Ascites |
|---|---|---|---|---|
| Low load-PEO1 cells | 0/3 | No | 14 | N/A |
| High load-PEO1 cells | 0/3 | No | 14 | N/A |
| Low load-PEO4 cells | 0/3 | No | 14 | N/A |
| High load-PEO4 cells | 3/3 | Yes | 6 | Bloody with MCS |
| Low load-PEO6 cells | 3/3 | Yes | 3.5 | Bloody with MCS |
| Location of Mets | PEO4 Cells | PEO6 Cells |
|---|---|---|
| Isolated masses | 1/3 | 3/6 |
| Omental-pancreatic area | 3/3 | 6/6 |
| Base of the liver | 2/3 | 3/6 |
| Diaphragm | 3/3 | 2/6 |
| Ovary-oviduct-uterine area | 3/3 | 5/6 |
| Peritoneal wall | 1/3 | 4/6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goyeneche, A.; Lisio, M.-A.; Fu, L.; Srinivasan, R.; Valdez Capuccino, J.; Gao, Z.-h.; Telleria, C. The Capacity of High-Grade Serous Ovarian Cancer Cells to Form Multicellular Structures Spontaneously along Disease Progression Correlates with Their Orthotopic Tumorigenicity in Immunosuppressed Mice. Cancers 2020, 12, 699. https://doi.org/10.3390/cancers12030699
Goyeneche A, Lisio M-A, Fu L, Srinivasan R, Valdez Capuccino J, Gao Z-h, Telleria C. The Capacity of High-Grade Serous Ovarian Cancer Cells to Form Multicellular Structures Spontaneously along Disease Progression Correlates with Their Orthotopic Tumorigenicity in Immunosuppressed Mice. Cancers. 2020; 12(3):699. https://doi.org/10.3390/cancers12030699
Chicago/Turabian StyleGoyeneche, Alicia, Michael-Anthony Lisio, Lili Fu, Rekha Srinivasan, Juan Valdez Capuccino, Zu-hua Gao, and Carlos Telleria. 2020. "The Capacity of High-Grade Serous Ovarian Cancer Cells to Form Multicellular Structures Spontaneously along Disease Progression Correlates with Their Orthotopic Tumorigenicity in Immunosuppressed Mice" Cancers 12, no. 3: 699. https://doi.org/10.3390/cancers12030699