Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting

 ,
,
Abstract
:1. Introduction
2. In Vivo Dendritic Cell Targeting
2.1. Fc Receptor Targeting
2.2. C-type Lectin Receptor Targeting
2.2.1. Mannose Receptor Targeting
2.2.2. DC-SIGN Receptor Targeting
2.2.3. DEC205 Receptor Targeting
2.2.4. Other C-Type Lectin Receptor Targeting
2.3. Scavenger Receptor Targeting
2.4. Targeting Dendritic Cells Using Genetically Modified Lymphocytes
2.5. Receptor-Mediated Maturation, Activation, and Migration of Dendritic Cells (Adjuvants)
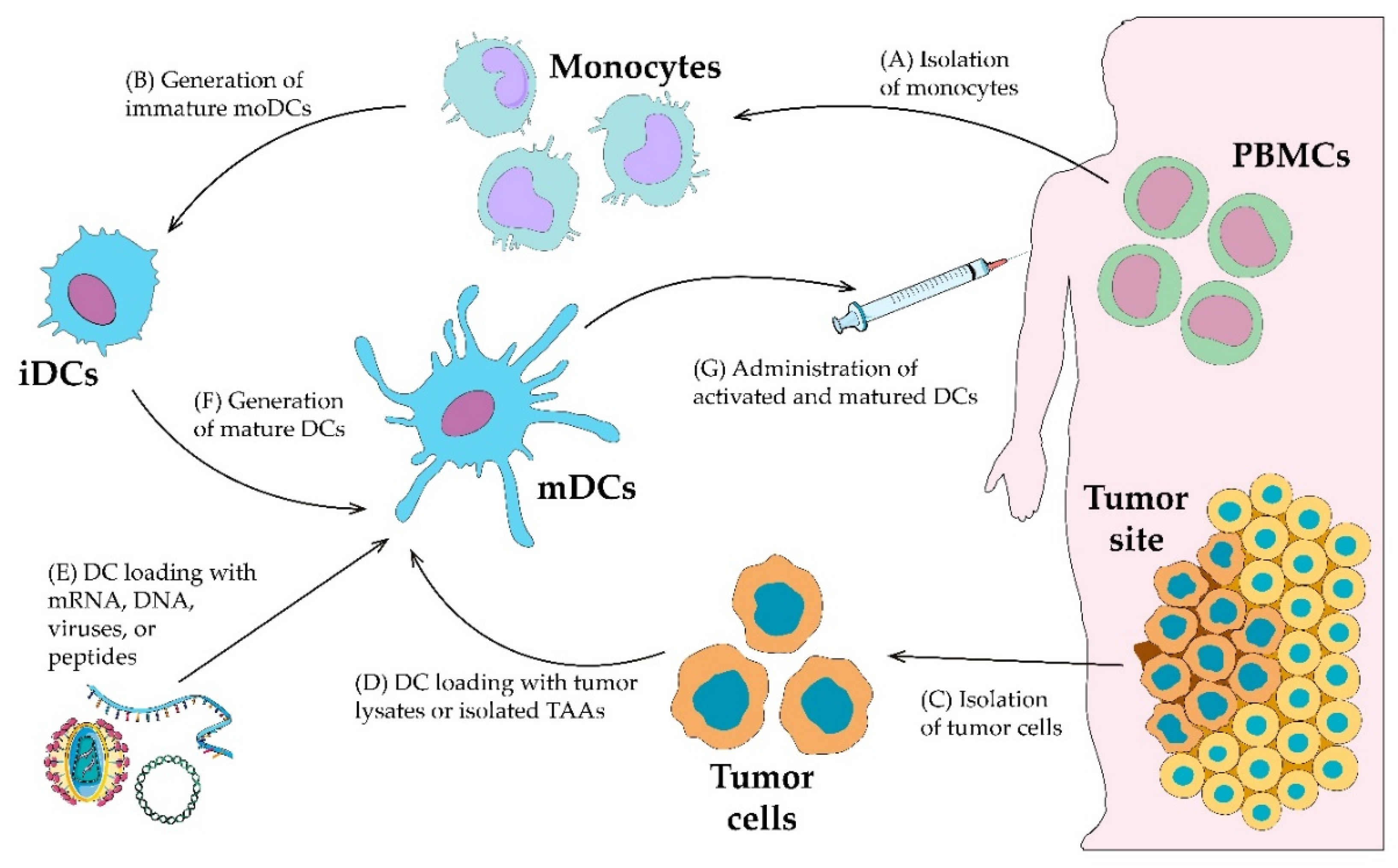
3. Ex Vivo Dendritic Cell Loading
3.1. Methods of Ex Vivo Dendritic Cell Generation
3.2. Dendritic Cell Loading with Peptides, Proteins and Tumor Cell Lysates
3.3. Dendritic Cell Transduction with Antigen-Encoding mRNA, DNA
3.4. Dendritic Cell Transduction with Viruses
3.5. Dendritic Cell Maturation/Stimulation Approaches
3.6. Dendritic/Tumor Fusion Cells
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Ann. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Thery, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Ann. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Lukas, D.; Clausen, B.E.; Yogev, N. Dendritic cells as gatekeepers of tolerance. Semin. Immunopathol. 2017, 39, 153–163. [Google Scholar] [CrossRef]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef]
- Fukaya, T.; Murakami, R.; Takagi, H.; Sato, K.; Sato, Y.; Otsuka, H.; Ohno, M.; Hijikata, A.; Ohara, O.; Hikida, M.; et al. Conditional ablation of CD205+ conventional dendritic cells impacts the regulation of T-cell immunity and homeostasis in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 11288–11293. [Google Scholar] [CrossRef] [Green Version]
- Mathan, T.S.; Figdor, C.G.; Buschow, S.I. Human plasmacytoid dendritic cells: From molecules to intercellular communication network. Front. Immunol. 2013, 4, 372. [Google Scholar] [CrossRef] [Green Version]
- Montoya, C.J.; Jie, H.B.; Al-Harthi, L.; Mulder, C.; Patino, P.J.; Rugeles, M.T.; Krieg, A.M.; Landay, A.L.; Wilson, S.B. Activation of plasmacytoid dendritic cells with TLR9 agonists initiates invariant NKT cell-mediated cross-talk with myeloid dendritic cells. J. Immunol. 2006, 177, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Brigl, M.; Tatituri, R.V.; Watts, G.F.; Bhowruth, V.; Leadbetter, E.A.; Barton, N.; Cohen, N.R.; Hsu, F.F.; Besra, G.S.; Brenner, M.B. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J. Exp. Med. 2011, 208, 1163–1177. [Google Scholar] [CrossRef]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of distinct natural killer cell subsets in anticancer response. Front. Immunol. 2017, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Barinov, A.; Galgano, A.; Krenn, G.; Tanchot, C.; Vasseur, F.; Rocha, B. CD4/CD8/Dendritic cell complexes in the spleen: CD8+ T cells can directly bind CD4+ T cells and modulate their response. PLoS ONE 2017, 12, e0180644. [Google Scholar] [CrossRef] [PubMed]
- Beuneu, H.; Garcia, Z.; Bousso, P. Cutting edge: Cognate CD4 help promotes recruitment of antigen-specific CD8 T cells around dendritic cells. J. Immunol. 2006, 177, 1406–1410. [Google Scholar] [CrossRef] [Green Version]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Delamarre, L.; Pack, M.; Chang, H.; Mellman, I.; Trombetta, E.S. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 2005, 307, 1630–1634. [Google Scholar] [CrossRef]
- Accapezzato, D.; Visco, V.; Francavilla, V.; Molette, C.; Donato, T.; Paroli, M.; Mondelli, M.U.; Doria, M.; Torrisi, M.R.; Barnaba, V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 2005, 202, 817–828. [Google Scholar] [CrossRef]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef] [Green Version]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Rodriguez, A.; Regnault, A.; Kleijmeer, M.; Ricciardi-Castagnoli, P.; Amigorena, S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999, 1, 362–368. [Google Scholar] [CrossRef]
- Jarvis, C.M.; Zwick, D.B.; Grim, J.C.; Alam, M.M.; Prost, L.R.; Gardiner, J.C.; Park, S.; Zimdars, L.L.; Sherer, N.M.; Kiessling, L.L. Antigen structure affects cellular routing through DC-SIGN. Proc. Natl. Acad. Sci. USA 2019, 116, 14862–14867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, X.Y.; Cascio, P.; Lemerise, K.; Goldberg, A.L.; Rock, K. Distinct proteolytic processes generate the C and N termini of MHC class I-binding peptides. J. Immunol. 1999, 163, 5851–5859. [Google Scholar] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, M.C.; Quintana, F.J. Tolerogenic dendritic cells. Semin. Immunopathol. 2017, 39, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Probst, H.C.; McCoy, K.; Okazaki, T.; Honjo, T.; van den Broek, M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 2005, 6, 280–286. [Google Scholar] [CrossRef]
- Schildknecht, A.; Brauer, S.; Brenner, C.; Lahl, K.; Schild, H.; Sparwasser, T.; Probst, H.C.; van den Broek, M. FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Yogev, N.; Frommer, F.; Lukas, D.; Kautz-Neu, K.; Karram, K.; Ielo, D.; von Stebut, E.; Probst, H.C.; van den Broek, M.; Riethmacher, D.; et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity 2012, 37, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Diversity and collaboration for effective immunotherapy. Nat. Med. 2016, 22, 1390–1391. [Google Scholar] [CrossRef]
- Zhu, E.F.; Gai, S.A.; Opel, C.F.; Kwan, B.H.; Surana, R.; Mihm, M.C.; Kauke, M.J.; Moynihan, K.D.; Angelini, A.; Williams, R.T.; et al. Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer Cell 2015, 27, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Moynihan, K.D.; Opel, C.F.; Szeto, G.L.; Tzeng, A.; Zhu, E.F.; Engreitz, J.M.; Williams, R.T.; Rakhra, K.; Zhang, M.H.; Rothschilds, A.M.; et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 2016, 22, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial watch: Dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, P.M.; Butterfield, L.H. Dendritic cell-based cancer vaccines. J. Immunol. 2018, 200, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, S.; Gentili, M.; Manel, N. Diversity of pathogen sensors in dendritic cells. Adv. Immunol. 2013, 120, 211–237. [Google Scholar] [CrossRef]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Cella, M.; Danieli, C.; Lanzavecchia, A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 1995, 182, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Reis e Sousa, C.; Stahl, P.D.; Austyn, J.M. Phagocytosis of antigens by Langerhans cells in vitro. J. Exp. Med. 1993, 178, 509–519. [Google Scholar] [CrossRef]
- Svensson, M.; Stockinger, B.; Wick, M.J. Bone marrow-derived dendritic cells can process bacteria for MHC-I and MHC-II presentation to T cells. J. Immunol. 1997, 158, 4229–4236. [Google Scholar]
- Theriault, J.R.; Adachi, H.; Calderwood, S.K. Role of scavenger receptors in the binding and internalization of heat shock protein 70. J. Immunol. 2006, 177, 8604–8611. [Google Scholar] [CrossRef] [Green Version]
- Tacken, P.J.; Figdor, C.G. Targeted antigen delivery and activation of dendritic cells in vivo: Steps towards cost effective vaccines. Semin. Immunol. 2011, 23, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G. Chemokines and the homing of dendritic cells to the T cell areas of lymphoid organs. J. Exp. Med. 1999, 189, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Angeli, V.; Swartz, M.A. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 2005, 5, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Chapman, R.; Powderly, J.; Blackwell, K.; Keler, T.; Green, J.; Riggs, R.; He, L.Z.; Ramakrishna, V.; Vitale, L.; et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4844–4853. [Google Scholar] [CrossRef] [Green Version]
- Somaiah, N.; Block, M.S.; Kim, J.W.; Shapiro, G.I.; Do, K.T.; Hwu, P.; Eder, J.P.; Jones, R.L.; Lu, H.; Ter Meulen, J.H.; et al. First-in-class, first-in-human study evaluating LV305, a dendritic-cell tropic lentiviral vector, in sarcoma and other solid tumors expressing NY-ESO-1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5808–5817. [Google Scholar] [CrossRef] [Green Version]
- Gargett, T.; Abbas, M.N.; Rolan, P.; Price, J.D.; Gosling, K.M.; Ferrante, A.; Ruszkiewicz, A.; Atmosukarto, I.I.C.; Altin, J.; Parish, C.R.; et al. Phase I trial of Lipovaxin-MM, a novel dendritic cell-targeted liposomal vaccine for malignant melanoma. Cancer Immunol. Immunother. CII 2018, 67, 1461–1472. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra51. [Google Scholar] [CrossRef]
- Maki, R.G.; Livingston, P.O.; Lewis, J.J.; Janetzki, S.; Klimstra, D.; Desantis, D.; Srivastava, P.K.; Brennan, M.F. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig. Dis. Sci. 2007, 52, 1964–1972. [Google Scholar] [CrossRef]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Lim, M.; Sughrue, M.E.; Komotar, R.J.; Abrahams, J.M.; O’Rourke, D.M.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A.T. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: Impact of peripheral PD-L1 expression on response to therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3575–3584. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro Oncol. 2014, 16, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Einstein, M.H.; Kadish, A.S.; Burk, R.D.; Kim, M.Y.; Wadler, S.; Streicher, H.; Goldberg, G.L.; Runowicz, C.D. Heat shock fusion protein-based immunotherapy for treatment of cervical intraepithelial neoplasia III. Gynecol. Oncol. 2007, 106, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorslaer, K.; Reimers, L.L.; Studentsov, Y.Y.; Einstein, M.H.; Burk, R.D. Serological response to an HPV16 E7 based therapeutic vaccine in women with high-grade cervical dysplasia. Gynecol. Oncol. 2010, 116, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilla, L.; Patuzzo, R.; Rivoltini, L.; Maio, M.; Pennacchioli, E.; Lamaj, E.; Maurichi, A.; Massarut, S.; Marchiano, A.; Santantonio, C.; et al. A phase II trial of vaccination with autologous, tumor-derived heat-shock protein peptide complexes Gp96, in combination with GM-CSF and interferon-alpha in metastatic melanoma patients. Cancer Immunol. Immunother. CII 2006, 55, 958–968. [Google Scholar] [CrossRef]
- Russo, V.; Pilla, L.; Lunghi, F.; Crocchiolo, R.; Greco, R.; Ciceri, F.; Maggioni, D.; Fontana, R.; Mukenge, S.; Rivoltini, L.; et al. Clinical and immunologic responses in melanoma patients vaccinated with MAGE-A3-genetically modified lymphocytes. Int. J. Cancer 2013, 132, 2557–2566. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting antigens to dendritic cell receptors for vaccine development. J. Drug Deliv. 2013, 2013, 869718. [Google Scholar] [CrossRef]
- Snider, D.P.; Kaubisch, A.; Segal, D.M. Enhanced antigen immunogenicity induced by bispecific antibodies. J. Exp. Med. 1990, 171, 1957–1963. [Google Scholar] [CrossRef] [Green Version]
- de Jong, J.M.; Schuurhuis, D.H.; Ioan-Facsinay, A.; van der Voort, E.I.; Huizinga, T.W.; Ossendorp, F.; Toes, R.E.; Verbeek, J.S. Murine Fc receptors for IgG are redundant in facilitating presentation of immune complex derived antigen to CD8+ T cells in vivo. Mol. Immunol. 2006, 43, 2045–2050. [Google Scholar] [CrossRef]
- Berlyn, K.A.; Schultes, B.; Leveugle, B.; Noujaim, A.A.; Alexander, R.B.; Mann, D.L. Generation of CD4(+) and CD8(+) T lymphocyte responses by dendritic cells armed with PSA/anti-PSA (antigen/antibody) complexes. Clin. Immunol. 2001, 101, 276–283. [Google Scholar] [CrossRef]
- Rhodes, J.W.; Tong, O.; Harman, A.N.; Turville, S.G. Human dendritic cell subsets, ontogeny, and impact on HIV infection. Front. Immunol. 2019, 10, 1088. [Google Scholar] [CrossRef] [Green Version]
- Schreibelt, G.; Klinkenberg, L.J.; Cruz, L.J.; Tacken, P.J.; Tel, J.; Kreutz, M.; Adema, G.J.; Brown, G.D.; Figdor, C.G.; de Vries, I.J. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012, 119, 2284–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Leeuwen-Kerkhoff, N.; Lundberg, K.; Westers, T.M.; Kordasti, S.; Bontkes, H.J.; de Gruijl, T.D.; Lindstedt, M.; van de Loosdrecht, A.A. Transcriptional profiling reveals functional dichotomy between human slan(+) non-classical monocytes and myeloid dendritic cells. J. Leukoc. Biol. 2017, 102, 1055–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzetti, F.; Tamassia, N.; Micheletti, A.; Finotti, G.; Bianchetto-Aguilera, F.; Cassatella, M.A. Human dendritic cell subset 4 (DC4) correlates to a subset of CD14(dim/-)CD16(++) monocytes. J. Allergy Clin. Immunol. 2018, 141, 2276–2279 e2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turville, S.G.; Cameron, P.U.; Handley, A.; Lin, G.; Pohlmann, S.; Doms, R.W.; Cunningham, A.L. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 2002, 3, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, M.T.; Loncaric, A.; Krutzik, S.R.; Becker, T.C.; Modlin, R.L. “Dermal dendritic cells” comprise two distinct populations: CD1+ dendritic cells and CD209+ macrophages. J. Investig. Dermatol. 2008, 128, 2225–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigley, V.; McGovern, N.; Milne, P.; Dickinson, R.; Pagan, S.; Cookson, S.; Haniffa, M.; Collin, M. Langerin-expressing dendritic cells in human tissues are related to CD1c+ dendritic cells and distinct from Langerhans cells and CD141high XCR1+ dendritic cells. J. Leukoc. Biol. 2015, 97, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Mommaas, M.; Oppel, T.; Schottdorf, E.M.; Gunther, S.; Moderer, M. Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J. Investig. Dermatol. 2002, 118, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulos, V.; McKenzie, I.F. Role of the mannose receptor in the immune response. Curr. Mol. Med. 2001, 1, 469–474. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Pietersz, G.A.; Gordon, S.; Martinez-Pomares, L.; McKenzie, I.F. Aldehyde-mannan antigen complexes target the MHC class I antigen-presentation pathway. Eur. J. Immunol. 2000, 30, 1714–1723. [Google Scholar] [CrossRef]
- Tan, M.C.; Mommaas, A.M.; Drijfhout, J.W.; Jordens, R.; Onderwater, J.J.; Verwoerd, D.; Mulder, A.A.; van der Heiden, A.N.; Scheidegger, D.; Oomen, L.C.; et al. Mannose receptor-mediated uptake of antigens strongly enhances HLA class II-restricted antigen presentation by cultured dendritic cells. Eur. J. Immunol. 1997, 27, 2426–2435. [Google Scholar] [CrossRef]
- Ramakrishna, V.; Treml, J.F.; Vitale, L.; Connolly, J.E.; O’Neill, T.; Smith, P.A.; Jones, C.L.; He, L.Z.; Goldstein, J.; Wallace, P.K.; et al. Mannose receptor targeting of tumor antigen pmel17 to human dendritic cells directs anti-melanoma T cell responses via multiple HLA molecules. J. Immunol. 2004, 172, 2845–2852. [Google Scholar] [CrossRef] [Green Version]
- He, L.Z.; Ramakrishna, V.; Connolly, J.E.; Wang, X.T.; Smith, P.A.; Jones, C.L.; Valkova-Valchanova, M.; Arunakumari, A.; Treml, J.F.; Goldstein, J.; et al. A novel human cancer vaccine elicits cellular responses to the tumor-associated antigen, human chorionic gonadotropin beta. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 1920–1927. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Matsuzaki, J.; Kelly, M.P.; Ramakrishna, V.; Vitale, L.; He, L.Z.; Keler, T.; Odunsi, K.; Old, L.J.; Ritter, G.; et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J. Immunol. 2011, 186, 1218–1227. [Google Scholar] [CrossRef] [Green Version]
- Avrameas, A.; McIlroy, D.; Hosmalin, A.; Autran, B.; Debre, P.; Monsigny, M.; Roche, A.C.; Midoux, P. Expression of a mannose/fucose membrane lectin on human dendritic cells. Eur. J. Immunol. 1996, 26, 394–400. [Google Scholar] [CrossRef]
- Wollenberg, A.; Kraft, S.; Hanau, D.; Bieber, T. Immunomorphological and ultrastructural characterization of Langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J. Investig. Dermatol. 1996, 106, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [Green Version]
- He, L.Z.; Crocker, A.; Lee, J.; Mendoza-Ramirez, J.; Wang, X.T.; Vitale, L.A.; O’Neill, T.; Petromilli, C.; Zhang, H.F.; Lopez, J.; et al. Antigenic targeting of the human mannose receptor induces tumor immunity. J. Immunol. 2007, 178, 6259–6267. [Google Scholar] [CrossRef] [Green Version]
- He, L.Z.; Weidlick, J.; Sisson, C.; Marsh, H.C.; Keler, T. Toll-like receptor agonists shape the immune responses to a mannose receptor-targeted cancer vaccine. Cell. Mol. Immunol. 2015, 12, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Garu, A.; Moku, G.; Gulla, S.K.; Chaudhuri, A. Genetic immunization with in vivo dendritic cell-targeting liposomal DNA vaccine carrier induces long-lasting antitumor immune response. Mol. Ther. 2016, 24, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Gulla, S.K.; Rao, B.R.; Moku, G.; Jinka, S.; Nimmu, N.V.; Khalid, S.; Patra, C.R.; Chaudhuri, A. In vivo targeting of DNA vaccines to dendritic cells using functionalized gold nanoparticles. Biomater. Sci. 2019, 7, 773–788. [Google Scholar] [CrossRef]
- Meka, R.R.; Mukherjee, S.; Patra, C.R.; Chaudhuri, A. Shikimoyl-ligand decorated gold nanoparticles for use in ex vivo engineered dendritic cell based DNA vaccination. Nanoscale 2019, 11, 7931–7943. [Google Scholar] [CrossRef]
- Raviv, L.; Jaron-Mendelson, M.; David, A. Mannosylated polyion complexes for in vivo gene delivery into CD11c(+) dendritic cells. Mol. Pharm. 2015, 12, 453–462. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.; Geijtenbeek, T.B.; van Kooyk, Y. Cutting edge: Carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef] [Green Version]
- Engering, A.; Geijtenbeek, T.B.; van Vliet, S.J.; Wijers, M.; van Liempt, E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef] [Green Version]
- McGovern, N.; Schlitzer, A.; Gunawan, M.; Jardine, L.; Shin, A.; Poyner, E.; Green, K.; Dickinson, R.; Wang, X.N.; Low, D.; et al. Human dermal CD14(+) cells are a transient population of monocyte-derived macrophages. Immunity 2014, 41, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, O.; Larrieu, P.; Duverger, E.; Boccaccio, C.; Bousser, M.T.; Monsigny, M.; Fonteneau, J.F.; Jotereau, F.; Roche, A.C. Synthesis of glycocluster-tumor antigenic peptide conjugates for dendritic cell targeting. Bioconjug. Chem. 2007, 18, 1547–1554. [Google Scholar] [CrossRef]
- Aarnoudse, C.A.; Bax, M.; Sanchez-Hernandez, M.; Garcia-Vallejo, J.J.; van Kooyk, Y. Glycan modification of the tumor antigen gp100 targets DC-SIGN to enhance dendritic cell induced antigen presentation to T cells. Int. J. Cancer 2008, 122, 839–846. [Google Scholar] [CrossRef]
- Arosio, D.; Chiodo, F.; Reina, J.J.; Marelli, M.; Penades, S.; van Kooyk, Y.; Garcia-Vallejo, J.J.; Bernardi, A. Effective targeting of DC-SIGN by alpha-fucosylamide functionalized gold nanoparticles. Bioconjug. Chem. 2014, 25, 2244–2251. [Google Scholar] [CrossRef] [Green Version]
- Berzi, A.; Ordanini, S.; Joosten, B.; Trabattoni, D.; Cambi, A.; Bernardi, A.; Clerici, M. Pseudo-Mannosylated DC-SIGN ligands as immunomodulants. Sci. Rep. 2016, 6, 35373. [Google Scholar] [CrossRef]
- Tacken, P.J.; de Vries, I.J.; Gijzen, K.; Joosten, B.; Wu, D.; Rother, R.P.; Faas, S.J.; Punt, C.J.; Torensma, R.; Adema, G.J.; et al. Effective induction of naive and recall T-cell responses by targeting antigen to human dendritic cells via a humanized anti-DC-SIGN antibody. Blood 2005, 106, 1278–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacken, P.J.; Ginter, W.; Berod, L.; Cruz, L.J.; Joosten, B.; Sparwasser, T.; Figdor, C.G.; Cambi, A. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 2011, 118, 4111–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, H.; Rideout, K.; Cho, T.; Joo, K.I.; Ziegler, L.; Elliot, A.; Walls, A.; Yu, D.; Baltimore, D.; et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat. Biotechnol. 2008, 26, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Tareen, S.U.; Kelley-Clarke, B.; Nicolai, C.J.; Cassiano, L.A.; Nelson, L.T.; Slough, M.M.; Vin, C.D.; Odegard, J.M.; Sloan, D.D.; Van Hoeven, N.; et al. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol. Ther. 2014, 22, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef]
- van Broekhoven, C.L.; Parish, C.R.; Demangel, C.; Britton, W.J.; Altin, J.G. Targeting dendritic cells with antigen-containing liposomes: A highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004, 64, 4357–4365. [Google Scholar] [CrossRef] [Green Version]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–684. [Google Scholar] [CrossRef]
- Kato, M.; McDonald, K.J.; Khan, S.; Ross, I.L.; Vuckovic, S.; Chen, K.; Munster, D.; MacDonald, K.P.; Hart, D.N. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int. Immunol. 2006, 18, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Shrimpton, R.E.; Butler, M.; Morel, A.S.; Eren, E.; Hue, S.S.; Ritter, M.A. CD205 (DEC-205): A recognition receptor for apoptotic and necrotic self. Mol. Immunol. 2009, 46, 1229–1239. [Google Scholar] [CrossRef]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.S.; Mahnke, K.; Storn, V.; Schonfeld, K.; Ring, S.; Nettelbeck, D.M.; Haisma, H.J.; Le Gall, F.; Kontermann, R.E.; Enk, A.H. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 8169–8177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Jin, Y.; Li, W.; Zhang, B.; He, Y.; Liu, H.; Xia, N.; Wei, H.; Yan, J. DNA vaccines targeting the encoded antigens to dendritic cells induce potent antitumor immunity in mice. BMC Immunol. 2013, 14, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Kuroiwa, J.M.; He, L.Z.; Charalambous, A.; Keler, T.; Steinman, R.M. The human cancer antigen mesothelin is more efficiently presented to the mouse immune system when targeted to the DEC-205/CD205 receptor on dendritic cells. Ann. N. Y. Acad. Sci. 2009, 1174, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Masilamani, R.F.; Bettelli, E.; Kuchroo, V.K.; Nussenzweig, M.C. Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 2004, 20, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Iberg, C.A.; Hawiger, D. Advancing immunomodulation by in vivo antigen delivery to DEC-205 and other cell surface molecules using recombinant chimeric antibodies. Int. Immunopharmacol. 2019, 73, 575–580. [Google Scholar] [CrossRef]
- Neubert, K.; Lehmann, C.H.; Heger, L.; Baranska, A.; Staedtler, A.M.; Buchholz, V.R.; Yamazaki, S.; Heidkamp, G.F.; Eissing, N.; Zebroski, H.; et al. Antigen delivery to CD11c+CD8- dendritic cells induces protective immune responses against experimental melanoma in mice in vivo. J. Immunol. 2014, 192, 5830–5838. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zaidi, N.; He, L.Z.; Zhang, L.; Kuroiwa, J.M.; Keler, T.; Steinman, R.M. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res. 2012, 14, R39. [Google Scholar] [CrossRef]
- Volckmar, J.; Knop, L.; Stegemann-Koniszewski, S.; Schulze, K.; Ebensen, T.; Guzman, C.A.; Bruder, D. The STING activator c-di-AMP exerts superior adjuvant properties than the formulation poly(I:C)/CpG after subcutaneous vaccination with soluble protein antigen or DEC-205-mediated antigen targeting to dendritic cells. Vaccine 2019, 37, 4963–4974. [Google Scholar] [CrossRef]
- Kreutz, M.; Giquel, B.; Hu, Q.; Abuknesha, R.; Uematsu, S.; Akira, S.; Nestle, F.O.; Diebold, S.S. Antibody-antigen-adjuvant conjugates enable co-delivery of antigen and adjuvant to dendritic cells in cis but only have partial targeting specificity. PLoS ONE 2012, 7, e40208. [Google Scholar] [CrossRef] [Green Version]
- Barbuto, S.; Idoyaga, J.; Vila-Perello, M.; Longhi, M.P.; Breton, G.; Steinman, R.M.; Muir, T.W. Induction of innate and adaptive immunity by delivery of poly dA:dT to dendritic cells. Nat. Chem. Biol. 2013, 9, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Pasqual, G.; Angelini, A.; Victora, G.D. Triggering positive selection of germinal center B cells by antigen targeting to DEC-205. Methods Mol. Biol. 2015, 1291, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Cheong, C.; Choi, J.H.; Vitale, L.; He, L.Z.; Trumpfheller, C.; Bozzacco, L.; Do, Y.; Nchinda, G.; Park, S.H.; Dandamudi, D.B.; et al. Improved cellular and humoral immune responses in vivo following targeting of HIV Gag to dendritic cells within human anti-human DEC205 monoclonal antibody. Blood 2010, 116, 3828–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dinther, D.; Stolk, D.A.; van de Ven, R.; van Kooyk, Y.; de Gruijl, T.D.; den Haan, J.M.M. Targeting C-type lectin receptors: A high-carbohydrate diet for dendritic cells to improve cancer vaccines. J. Leukoc. Biol. 2017, 102, 1017–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.; Rizzitelli, A.; Wu, L.; Vremec, D.; van Dommelen, S.L.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Robbins, S.H.; Walzer, T.; Dembele, D.; Thibault, C.; Defays, A.; Bessou, G.; Xu, H.; Vivier, E.; Sellars, M.; Pierre, P.; et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol. 2008, 9, R17. [Google Scholar] [CrossRef]
- Huysamen, C.; Willment, J.A.; Dennehy, K.M.; Brown, G.D. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 2008, 283, 16693–16701. [Google Scholar] [CrossRef] [Green Version]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Hochrein, H.; Shortman, K.; Vremec, D.; Scott, B.; Hertzog, P.; O’Keeffe, M. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 2001, 166, 5448–5455. [Google Scholar] [CrossRef] [Green Version]
- Maldonado-Lopez, R.; De Smedt, T.; Pajak, B.; Heirman, C.; Thielemans, K.; Leo, O.; Urbain, J.; Maliszewski, C.R.; Moser, M. Role of CD8alpha+ and CD8alpha-dendritic cells in the induction of primary immune responses in vivo. J. Leukoc. Biol. 1999, 66, 242–246. [Google Scholar] [CrossRef]
- Picco, G.; Beatson, R.; Taylor-Papadimitriou, J.; Burchell, J.M. Targeting DNGR-1 (CLEC9A) with antibody/MUC1 peptide conjugates as a vaccine for carcinomas. Eur. J. Immunol. 2014, 44, 1947–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 2011, 187, 842–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ahmet, F.; Sullivan, L.C.; Brooks, A.G.; Kent, S.J.; De Rose, R.; Salazar, A.M.; Reis e Sousa, C.; Shortman, K.; Lahoud, M.H.; et al. Antibodies targeting Clec9A promote strong humoral immunity without adjuvant in mice and non-human primates. Eur. J. Immunol. 2015, 45, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Tan, P.S.; Kavishna, R.; Ker, A.; Lu, J.; Chan, C.E.Z.; Hanson, B.J.; MacAry, P.A.; Caminschi, I.; Shortman, K.; et al. Enhancing vaccine antibody responses by targeting Clec9A on dendritic cells. NPJ Vaccin. 2017, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Guttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Tullett, K.M.; Leal Rojas, I.M.; Minoda, Y.; Tan, P.S.; Zhang, J.G.; Smith, C.; Khanna, R.; Shortman, K.; Caminschi, I.; Lahoud, M.H.; et al. Targeting CLEC9A delivers antigen to human CD141(+) DC for CD4(+) and CD8(+)T cell recognition. JCI Insight 2016, 1, e87102. [Google Scholar] [CrossRef]
- Yan, Z.; Wu, Y.; Du, J.; Li, G.; Wang, S.; Cao, W.; Zhou, X.; Wu, C.; Zhang, D.; Jing, X.; et al. A novel peptide targeting Clec9a on dendritic cell for cancer immunotherapy. Oncotarget 2016, 7, 40437–40450. [Google Scholar] [CrossRef] [Green Version]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martinez, D.; Hernanz-Falcon, P.; Rosewell, I.; Reis e Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanc, P.; Kjaer, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Middelberg, A.P.; Gemiarto, A.; MacDonald, K.; Baxter, A.G.; Talekar, M.; Moi, D.; Tullett, K.M.; Caminschi, I.; Lahoud, M.H.; et al. Self-adjuvanting nanoemulsion targeting dendritic cell receptor Clec9A enables antigen-specific immunotherapy. J. Clin. Investig. 2018, 128, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Bates, E.E.; Fournier, N.; Garcia, E.; Valladeau, J.; Durand, I.; Pin, J.J.; Zurawski, S.M.; Patel, S.; Abrams, J.S.; Lebecque, S.; et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J. Immunol. 1999, 163, 1973–1983. [Google Scholar] [PubMed]
- Lee, R.T.; Hsu, T.L.; Huang, S.K.; Hsieh, S.L.; Wong, C.H.; Lee, Y.C. Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology 2011, 21, 512–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klechevsky, E.; Flamar, A.L.; Cao, Y.; Blanck, J.P.; Liu, M.; O’Bar, A.; Agouna-Deciat, O.; Klucar, P.; Thompson-Snipes, L.; Zurawski, S.; et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood 2010, 116, 1685–1697. [Google Scholar] [CrossRef]
- Meyer-Wentrup, F.; Benitez-Ribas, D.; Tacken, P.J.; Punt, C.J.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood 2008, 111, 4245–4253. [Google Scholar] [CrossRef] [Green Version]
- Bloem, K.; Vuist, I.M.; van der Plas, A.J.; Knippels, L.M.; Garssen, J.; Garcia-Vallejo, J.J.; van Vliet, S.J.; van Kooyk, Y. Ligand binding and signaling of dendritic cell immunoreceptor (DCIR) is modulated by the glycosylation of the carbohydrate recognition domain. PLoS ONE 2013, 8, e66266. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Wentrup, F.; Cambi, A.; Joosten, B.; Looman, M.W.; de Vries, I.J.; Figdor, C.G.; Adema, G.J. DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J. Leukoc. Biol. 2009, 85, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Gunther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef]
- Hutten, T.J.; Thordardottir, S.; Fredrix, H.; Janssen, L.; Woestenenk, R.; Tel, J.; Joosten, B.; Cambi, A.; Heemskerk, M.H.; Franssen, G.M.; et al. CLEC12A-Mediated Antigen Uptake and Cross-Presentation by Human Dendritic Cell Subsets Efficiently Boost Tumor-Reactive T Cell Responses. J. Immunol. 2016, 197, 2715–2725. [Google Scholar] [CrossRef]
- Marshall, A.S.; Willment, J.A.; Lin, H.H.; Williams, D.L.; Gordon, S.; Brown, G.D. Identification and characterization of a novel human myeloid inhibitory C-type lectin-like receptor (MICL) that is predominantly expressed on granulocytes and monocytes. J. Biol. Chem. 2004, 279, 14792–14802. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, M.; Li, N.; Chen, T.; Zhang, Y.; Wan, T.; Cao, X. KLRL1, a novel killer cell lectinlike receptor, inhibits natural killer cell cytotoxicity. Blood 2004, 104, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Eidsmo, L.; Wu, L.; Sathe, P.; Pietersz, S.; Chang, H.W.; Walker, I.D.; et al. The C-type lectin Clec12A present on mouse and human dendritic cells can serve as a target for antigen delivery and enhancement of antibody responses. J. Immunol. 2009, 182, 7587–7594. [Google Scholar] [CrossRef]
- Murphy, J.E.; Tedbury, P.R.; Homer-Vanniasinkam, S.; Walker, J.H.; Ponnambalam, S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 2005, 182, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bansal, P.; Mukherjee, P.; Basu, S.K.; George, A.; Bal, V.; Rath, S. MHC class I-restricted presentation of maleylated protein binding to scavenger receptors. J. Immunol. 1999, 162, 4430–4437. [Google Scholar] [PubMed]
- Vandaveer, S.S.; Erf, G.F.; Durdik, J.M. Avian T helper one/two immune response balance can be shifted toward inflammation by antigen delivery to scavenger receptors. Poult. Sci. 2001, 80, 172–181. [Google Scholar] [CrossRef]
- Kissick, H.T.; Dunn, L.K.; Ghosh, S.; Nechama, M.; Kobzik, L.; Arredouani, M.S. The scavenger receptor MARCO modulates TLR-induced responses in dendritic cells. PLoS ONE 2014, 9, e104148. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Varin, A.; Chen, Y.; Liu, B.; Tryggvason, K.; Gordon, S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 2011, 117, 1319–1328. [Google Scholar] [CrossRef]
- Abraham, R.; Singh, N.; Mukhopadhyay, A.; Basu, S.K.; Bal, V.; Rath, S. Modulation of immunogenicity and antigenicity of proteins by maleylation to target scavenger receptors on macrophages. J. Immunol. 1995, 154, 1–8. [Google Scholar]
- Yin, W.; Gorvel, L.; Zurawski, S.; Li, D.; Ni, L.; Duluc, D.; Upchurch, K.; Kim, J.; Gu, C.; Ouedraogo, R.; et al. Functional Specialty of CD40 and Dendritic Cell Surface Lectins for Exogenous Antigen Presentation to CD8(+) and CD4(+) T Cells. EBioMedicine 2016, 5, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, J.; Ding, L.; Jin, H.; Lovell, J.F.; Corbin, I.R.; Cao, W.; Lo, P.C.; Yang, M.; Tsao, M.S.; et al. HDL-mimicking peptide-lipid nanoparticles with improved tumor targeting. Small 2010, 6, 430–437. [Google Scholar] [CrossRef]
- Qian, Y.; Jin, H.; Qiao, S.; Dai, Y.; Huang, C.; Lu, L.; Luo, Q.; Zhang, Z. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials 2016, 98, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Protein misassembly: Macromolecular crowding and molecular chaperones. Adv. Exp. Med. Biol. 2007, 594, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Binder, R.J.; Suto, R.; Anderson, K.M.; Srivastava, P.K. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 2000, 12, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Febbraio, M.A. Exosome-dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blachere, N.E.; Li, Z.; Chandawarkar, R.Y.; Suto, R.; Jaikaria, N.S.; Basu, S.; Udono, H.; Srivastava, P.K. Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J. Exp. Med. 1997, 186, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Gong, J.; Calderwood, S.K. Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology 2014, 219, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Baldin, A.V.; Zamyatnin, A.A., Jr.; Bazhin, A.V.; Xu, W.H.; Savvateeva, L.V. Advances in the Development of Anticancer HSP-based Vaccines. Curr. Med. Chem. 2019, 26, 427–445. [Google Scholar] [CrossRef]
- Tamura, Y.; Peng, P.; Liu, K.; Daou, M.; Srivastava, P.K. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science 1997, 278, 117–120. [Google Scholar] [CrossRef]
- Staib, F.; Distler, M.; Bethke, K.; Schmitt, U.; Galle, P.R.; Heike, M. Cross-presentation of human melanoma peptide antigen MART-1 to CTLs from in vitro reconstituted gp96/MART-1 complexes. Cancer Immun. 2004, 4, 3. [Google Scholar]
- Savvateeva, L.V.; Schwartz, A.M.; Gorshkova, L.B.; Gorokhovets, N.V.; Makarov, V.A.; Reddy, V.P.; Aliev, G.; Zamyatnin, A.A., Jr. Prophylactic Admission of an In Vitro Reconstructed Complexes of Human Recombinant Heat Shock Proteins and Melanoma Antigenic Peptides Activates Anti-Melanoma Responses in Mice. Curr. Mol. Med. 2015, 15, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, S.; Nishikawa, M.; Guan, X.; Ohno, Y.; Yata, T.; Takakura, Y. Enhanced generation of cytotoxic T lymphocytes by heat shock protein 70 fusion proteins harboring both CD8(+) T cell and CD4(+) T cell epitopes. Mol. Pharm. 2010, 7, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Iezzi, G.; Scheidegger, D.; Lanzavecchia, A. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 2001, 193, 987–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton, R.W.; Bradley, L.M.; Swain, S.L. T cell memory. Annu. Rev. Immunol. 1998, 16, 201–223. [Google Scholar] [CrossRef]
- Berger, C.; Flowers, M.E.; Warren, E.H.; Riddell, S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006, 107, 2294–2302. [Google Scholar] [CrossRef] [Green Version]
- Russo, V.; Cipponi, A.; Raccosta, L.; Rainelli, C.; Fontana, R.; Maggioni, D.; Lunghi, F.; Mukenge, S.; Ciceri, F.; Bregni, M.; et al. Lymphocytes genetically modified to express tumor antigens target DCs in vivo and induce antitumor immunity. J. Clin. Investig. 2007, 117, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Fontana, R.; Bregni, M.; Cipponi, A.; Raccosta, L.; Rainelli, C.; Maggioni, D.; Lunghi, F.; Ciceri, F.; Mukenge, S.; Doglioni, C.; et al. Peripheral blood lymphocytes genetically modified to express the self/tumor antigen MAGE-A3 induce antitumor immune responses in cancer patients. Blood 2009, 113, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Reis e Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Ebersold, M.; Garrett, W.; Pypaert, M.; Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 2003, 299, 1400–1403. [Google Scholar] [CrossRef]
- Delamarre, L.; Holcombe, H.; Mellman, I. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J. Exp. Med. 2003, 198, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Gil-Torregrosa, B.C.; Lennon-Dumenil, A.M.; Kessler, B.; Guermonprez, P.; Ploegh, H.L.; Fruci, D.; van Endert, P.; Amigorena, S. Control of cross-presentation during dendritic cell maturation. Eur. J. Immunol. 2004, 34, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Garbi, N.; Hammerling, G.J.; Probst, H.C.; van den Broek, M. Tonic T cell signalling and T cell tolerance as opposite effects of self-recognition on dendritic cells. Curr. Opin. Immunol. 2010, 22, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Hochweller, K.; Wabnitz, G.H.; Samstag, Y.; Suffner, J.; Hammerling, G.J.; Garbi, N. Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proc. Natl. Acad. Sci. USA 2010, 107, 5931–5936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Iyoda, T.; Tarbell, K.; Olson, K.; Velinzon, K.; Inaba, K.; Steinman, R.M. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 2003, 198, 235–247. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Arnold-Schrauf, C.; Berod, L.; Sparwasser, T. Dendritic cell specific targeting of MyD88 signalling pathways in vivo. Eur. J. Immunol. 2015, 45, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Dutcher, J.P.; Anderson, J.E.; Eckhardt, S.G.; Stephans, K.F.; Razvillas, B.; Garl, S.; Butine, M.D.; Perry, V.P.; Armitage, R.J.; et al. Phase I study of recombinant human CD40 ligand in cancer patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 3280–3287. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef]
- Advani, R.; Forero-Torres, A.; Furman, R.R.; Rosenblatt, J.D.; Younes, A.; Ren, H.; Harrop, K.; Whiting, N.; Drachman, J.G. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4371–4377. [Google Scholar] [CrossRef]
- Wei, X.X.; Chan, S.; Kwek, S.; Lewis, J.; Dao, V.; Zhang, L.; Cooperberg, M.R.; Ryan, C.J.; Lin, A.M.; Friedlander, T.W.; et al. Systemic GM-CSF recruits effector T cells into the tumor microenvironment in localized prostate cancer. Cancer Immunol. Res. 2016, 4, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Hori, S.; Ohnishi, S.; Toritsuka, M.; Fujii, T.; Shimizu, T.; Owari, T.; Morizawa, Y.; Gotoh, D.; Itami, Y.; et al. Supplementary granulocyte macrophage colony-stimulating factor to chemotherapy and programmed death-ligand 1 blockade decreases local recurrence after surgery in bladder cancer. Cancer Sci. 2019, 110, 3315–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessureault, S.; Alsarraj, M.; McCarthy, S.; Hunter, T.; Noyes, D.; Lee, D.; Harkins, J.; Seigne, J.; Jennings, R.; Antonia, S.J. A GM-CSF/CD40L producing cell augments anti-tumor T cell responses. J. Surg. Res. 2005, 125, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.; Mediavilla-Varela, M.; Antonia, S.J. A GM-CSF and CD40L bystander vaccine is effective in a murine breast cancer model. Breast Cancer (Dove Med. Press) 2015, 7, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessureault, S.; Noyes, D.; Lee, D.; Dunn, M.; Janssen, W.; Cantor, A.; Sotomayor, E.; Messina, J.; Antonia, S.J. A phase-I trial using a universal GM-CSF-producing and CD40L-expressing bystander cell line (GM.CD40L) in the formulation of autologous tumor cell-based vaccines for cancer patients with stage IV disease. Ann. Surg. Oncol. 2007, 14, 869–884. [Google Scholar] [CrossRef]
- Gray, J.E.; Chiappori, A.; Williams, C.C.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.C.; Kim, J.; Boyle, T.A.; Pinder-Schenck, M.; Khalil, F.; et al. A phase I/randomized phase II study of GM.CD40L vaccine in combination with CCL21 in patients with advanced lung adenocarcinoma. Cancer Immunol. Immunother. CII 2018, 67, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Antonia, S.; Noyes, D.; Hunter, T.B.; Simon, G.R.; Bepler, G.; Williams, C.C.; Tanvetyanon, T.; Haura, E.B.; Schell, M.J.; et al. Phase II trial of a GM-CSF-producing and CD40L-expressing bystander cell line combined with an allogeneic tumor cell-based vaccine for refractory lung adenocarcinoma. J. Immunother. 2013, 36, 442–450. [Google Scholar] [CrossRef] [Green Version]
- Murshid, A.; Borges, T.J.; Calderwood, S.K. Emerging roles for scavenger receptor SREC-I in immunity. Cytokine 2015, 75, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Beauvillain, C.; Meloni, F.; Sirard, J.C.; Blanchard, S.; Jarry, U.; Scotet, M.; Magistrelli, G.; Delneste, Y.; Barnaba, V.; Jeannin, P. The scavenger receptors SRA-1 and SREC-I cooperate with TLR2 in the recognition of the hepatitis C virus non-structural protein 3 by dendritic cells. J. Hepatol. 2010, 52, 644–651. [Google Scholar] [CrossRef]
- Dong, X.; Liang, J.; Yang, A.; Qian, Z.; Kong, D.; Lv, F. A visible codelivery nanovaccine of antigen and adjuvant with self-carrier for cancer immunotherapy. ACS Appl. Mater. Interfaces 2019, 11, 4876–4888. [Google Scholar] [CrossRef]
- Garg, A.D.; Vara Perez, M.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology 2017, 6, e1328341. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, D.; Bhardwaj, N. Generation of autologous peptide- and protein-pulsed dendritic cells for patient-specific immunotherapy. Methods Mol. Med. 2005, 109, 97–112. [Google Scholar] [PubMed]
- Fields, R.C.; Shimizu, K.; Mule, J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 9482–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizee, G.; Gonzales, M.I.; Topalian, S.L. Lentivirus vector-mediated expression of tumor-associated epitopes by human antigen presenting cells. Hum. Gene Ther. 2004, 15, 393–404. [Google Scholar] [CrossRef]
- Gilboa, E.; Vieweg, J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol. Rev. 2004, 199, 251–263. [Google Scholar] [CrossRef]
- Kajihara, M.; Takakura, K.; Ohkusa, T.; Koido, S. The impact of dendritic cell-tumor fusion cells on cancer vaccines - past progress and future strategies. Immunotherapy 2015, 7, 1111–1122. [Google Scholar] [CrossRef]
- Thurner, B.; Roder, C.; Dieckmann, D.; Heuer, M.; Kruse, M.; Glaser, A.; Keikavoussi, P.; Kampgen, E.; Bender, A.; Schuler, G. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J. Immunol. Methods 1999, 223, 1–15. [Google Scholar] [CrossRef]
- Bender, A.; Sapp, M.; Schuler, G.; Steinman, R.M.; Bhardwaj, N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods 1996, 196, 121–135. [Google Scholar] [CrossRef]
- Balan, S.; Dalod, M. In vitro generation of human XCR1(+) dendritic cells from CD34(+) hematopoietic progenitors. Methods Mol. Biol. 2016, 1423, 19–37. [Google Scholar] [CrossRef]
- Proietto, A.I.; Mittag, D.; Roberts, A.W.; Sprigg, N.; Wu, L. The equivalents of human blood and spleen dendritic cell subtypes can be generated in vitro from human CD34(+) stem cells in the presence of fms-like tyrosine kinase 3 ligand and thrombopoietin. Cell. Mol. Immunol. 2012, 9, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Thordardottir, S.; Hangalapura, B.N.; Hutten, T.; Cossu, M.; Spanholtz, J.; Schaap, N.; Radstake, T.R.; van der Voort, R.; Dolstra, H. The aryl hydrocarbon receptor antagonist StemRegenin 1 promotes human plasmacytoid and myeloid dendritic cell development from CD34+ hematopoietic progenitor cells. Stem Cells Dev. 2014, 23, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.; Gomes, C.; Falcao, A.; Neves, B.M.; Cruz, M.T. Dendritic cell-based immunotherapy: A basic review and recent advances. Immunol. Res. 2017, 65, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Mortarini, R.; Baldassari, P.; Guidetti, A.; Gallino, G.F.; Del Vecchio, M.; Ravagnani, F.; Magni, M.; Chaplin, P.; et al. Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD34(+)-derived dendritic cell vaccination: A phase I trial in metastatic melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 5381–5390. [Google Scholar] [CrossRef] [Green Version]
- Mackensen, A.; Herbst, B.; Chen, J.L.; Kohler, G.; Noppen, C.; Herr, W.; Spagnoli, G.C.; Cerundolo, V.; Lindemann, A. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int. J. Cancer 2000, 86, 385–392. [Google Scholar] [CrossRef]
- Banchereau, J.; Palucka, A.K.; Dhodapkar, M.; Burkeholder, S.; Taquet, N.; Rolland, A.; Taquet, S.; Coquery, S.; Wittkowski, K.M.; Bhardwaj, N.; et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001, 61, 6451–6458. [Google Scholar]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Bhardwaj, N. Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Santegoets, S.J.; van den Eertwegh, A.J.; van de Loosdrecht, A.A.; Scheper, R.J.; de Gruijl, T.D. Human dendritic cell line models for DC differentiation and clinical DC vaccination studies. J. Leukoc. Biol. 2008, 84, 1364–1373. [Google Scholar] [CrossRef] [Green Version]
- Masterson, A.J.; Sombroek, C.C.; De Gruijl, T.D.; Graus, Y.M.; van der Vliet, H.J.; Lougheed, S.M.; van den Eertwegh, A.J.; Pinedo, H.M.; Scheper, R.J. MUTZ-3, a human cell line model for the cytokine-induced differentiation of dendritic cells from CD34+ precursors. Blood 2002, 100, 701–703. [Google Scholar] [CrossRef] [Green Version]
- van Helden, S.F.; van Leeuwen, F.N.; Figdor, C.G. Human and murine model cell lines for dendritic cell biology evaluated. Immunol. Lett. 2008, 117, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.; Schreurs, M.W.; Masterson, A.J.; Liu, Y.P.; Goletz, S.; Baumeister, H.; Kueter, E.W.; Lougheed, S.M.; van den Eertwegh, A.J.; Scheper, R.J.; et al. In vitro priming of tumor-specific cytotoxic T lymphocytes using allogeneic dendritic cells derived from the human MUTZ-3 cell line. Cancer Immunol. Immunother. CII 2006, 55, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Kale, V.; Limaye, L. Umbilical cord blood-derived CD11c(+) dendritic cells could serve as an alternative allogeneic source of dendritic cells for cancer immunotherapy. Stem Cell Res. Ther. 2015, 6, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. CII 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, H.; Jiang, Y.; Gallo, R.C.; Cheng, H. Induction of antitumor cytotoxic lymphocytes using engineered human primary blood dendritic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E4453–E4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescigno, M.; Martino, M.; Sutherland, C.L.; Gold, M.R.; Ricciardi-Castagnoli, P. Dendritic cell survival and maturation are regulated by different signaling pathways. J. Exp. Med. 1998, 188, 2175–2180. [Google Scholar] [CrossRef]
- Ardeshna, K.M.; Pizzey, A.R.; Devereux, S.; Khwaja, A. The PI3 kinase, p38 SAP kinase, and NF-kappaB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood 2000, 96, 1039–1046. [Google Scholar] [CrossRef]
- Reitsma, D.J.; Combest, A.J. Challenges in the development of an autologous heat shock protein based anti-tumor vaccine. Hum. Vaccin. Immunother. 2012, 8, 1152–1155. [Google Scholar] [CrossRef] [Green Version]
- Moeller, I.; Spagnoli, G.C.; Finke, J.; Veelken, H.; Houet, L. Uptake routes of tumor-antigen MAGE-A3 by dendritic cells determine priming of naive T-cell subtypes. Cancer Immunol. Immunother. CII 2012, 61, 2079–2090. [Google Scholar] [CrossRef]
- Schnurr, M.; Chen, Q.; Shin, A.; Chen, W.; Toy, T.; Jenderek, C.; Green, S.; Miloradovic, L.; Drane, D.; Davis, I.D.; et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood 2005, 105, 2465–2472. [Google Scholar] [CrossRef]
- Nair, S.K.; Morse, M.; Boczkowski, D.; Cumming, R.I.; Vasovic, L.; Gilboa, E.; Lyerly, H.K. Induction of tumor-specific cytotoxic T lymphocytes in cancer patients by autologous tumor RNA-transfected dendritic cells. Ann. Surg. 2002, 235, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.R.; Tsakou, G.; Grunebach, F.; Schmidt, S.M.; Brossart, P. Induction of chronic lymphocytic leukemia (CLL)-specific CD4- and CD8-mediated T-cell responses using RNA-transfected dendritic cells. Blood 2004, 103, 1763–1769. [Google Scholar] [CrossRef] [Green Version]
- Borch, T.H.; Svane, I.M.; Met, O. Immune monitoring using mRNA-transfected dendritic cells. Methods Mol. Biol. 2016, 1428, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Heiser, A.; Maurice, M.A.; Yancey, D.R.; Coleman, D.M.; Dahm, P.; Vieweg, J. Human dendritic cells transfected with renal tumor RNA stimulate polyclonal T-cell responses against antigens expressed by primary and metastatic tumors. Cancer Res. 2001, 61, 3388–3393. [Google Scholar] [PubMed]
- Koido, S.; Kashiwaba, M.; Chen, D.; Gendler, S.; Kufe, D.; Gong, J. Induction of antitumor immunity by vaccination of dendritic cells transfected with MUC1 RNA. J. Immunol. 2000, 165, 5713–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, S.; Wilgenhof, S.; Heirman, C.; Corthals, J.; Breckpot, K.; Bonehill, A.; Neyns, B.; Thielemans, K. Optimized dendritic cell-based immunotherapy for melanoma: The TriMix-formula. Cancer Immunol. Immunother. CII 2014, 63, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Van Nuffel, A.M.; Wilgenhof, S.; Bonehill, A. Single-step antigen loading and maturation of dendritic cells through mRNA electroporation of a tumor-associated antigen and a TriMix of costimulatory molecules. Methods Mol. Biol. 2014, 1139, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pen, J.J.; De Keersmaecker, B.; Maenhout, S.K.; Van Nuffel, A.M.; Heirman, C.; Corthals, J.; Escors, D.; Bonehill, A.; Thielemans, K.; Breckpot, K.; et al. Modulation of regulatory T cell function by monocyte-derived dendritic cells matured through electroporation with mRNA encoding CD40 ligand, constitutively active TLR4, and CD70. J. Immunol. 2013, 191, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. CII 2015, 64, 381–388. [Google Scholar] [CrossRef]
- Voshavar, C.; Meka, R.C.; Samanta, S.; Marepally, S.; Chaudhuri, A. Enhanced spacer length between mannose mimicking shikimoyl and quinoyl headgroups and hydrophobic region of cationic amphiphile increases efficiency of dendritic cell based DNA vaccination: A structure-activity investigation. J. Med. Chem. 2017, 60, 1605–1610. [Google Scholar] [CrossRef]
- Sabado, R.L.; Bhardwaj, N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy 2010, 2, 37–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, L.H.; Comin-Anduix, B.; Vujanovic, L.; Lee, Y.; Dissette, V.B.; Yang, J.Q.; Vu, H.T.; Seja, E.; Oseguera, D.K.; Potter, D.M.; et al. Adenovirus MART-1-engineered autologous dendritic cell vaccine for metastatic melanoma. J. Immunother. 2008, 31, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroers, R.; Sinha, I.; Segall, H.; Schmidt-Wolf, I.G.; Rooney, C.M.; Brenner, M.K.; Sutton, R.E.; Chen, S.Y. Transduction of human PBMC-derived dendritic cells and macrophages by an HIV-1-based lentiviral vector system. Mol. Ther. J. Am. Soc. Gene Ther. 2000, 1, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Dyall, J.; Latouche, J.B.; Schnell, S.; Sadelain, M. Lentivirus-transduced human monocyte-derived dendritic cells efficiently stimulate antigen-specific cytotoxic T lymphocytes. Blood 2001, 97, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Breckpot, K.; Heirman, C.; De Greef, C.; van der Bruggen, P.; Thielemans, K. Identification of new antigenic peptide presented by HLA-Cw7 and encoded by several MAGE genes using dendritic cells transduced with lentiviruses. J. Immunol. 2004, 172, 2232–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhang, J.; Mi, Z.; Robbins, P.; Falo, L.D., Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J. Immunol. 2005, 174, 3808–3817. [Google Scholar] [CrossRef] [Green Version]
- Sundarasetty, B.S.; Chan, L.; Darling, D.; Giunti, G.; Farzaneh, F.; Schenck, F.; Naundorf, S.; Kuehlcke, K.; Ruggiero, E.; Schmidt, M.; et al. Lentivirus-induced ‘Smart’ dendritic cells: Pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Ther. 2015, 22, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Nicolette, C.A.; Healey, D.; Tcherepanova, I.; Whelton, P.; Monesmith, T.; Coombs, L.; Finke, L.H.; Whiteside, T.; Miesowicz, F. Dendritic cells for active immunotherapy: Optimizing design and manufacture in order to develop commercially and clinically viable products. Vaccine 2007, 25, B47–B60. [Google Scholar] [CrossRef]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef]
- Koido, S.; Homma, S.; Hara, E.; Mitsunaga, M.; Namiki, Y.; Takahara, A.; Nagasaki, E.; Komita, H.; Sagawa, Y.; Ohkusa, T.; et al. In vitro generation of cytotoxic and regulatory T cells by fusions of human dendritic cells and hepatocellular carcinoma cells. J. Transl. Med. 2008, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Inaba, K.; Pack, M.; Inaba, M.; Sakuta, H.; Isdell, F.; Steinman, R.M. High levels of a major histocompatibility complex II-self peptide complex on dendritic cells from the T cell areas of lymph nodes. J. Exp. Med. 1997, 186, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koido, S.; Ohana, M.; Liu, C.; Nikrui, N.; Durfee, J.; Lerner, A.; Gong, J. Dendritic cells fused with human cancer cells: Morphology, antigen expression, and T cell stimulation. Clin. Immunol. 2004, 113, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Gong, J. Characterization of structure and direct antigen presentation by dendritic/tumor-fused cells as cancer vaccines. Anticancer Res. 2013, 33, 347–354. [Google Scholar] [PubMed]
- Koido, S.; Enomoto, Y.; Apostolopoulos, V.; Gong, J. Tumor regression by CD4 T-cells primed with dendritic/tumor fusion cell vaccines. Anticancer Res. 2014, 34, 3917–3924. [Google Scholar] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Fusions between dendritic cells and whole tumor cells as anticancer vaccines. Oncoimmunology 2013, 2, e24437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koido, S.; Hara, E.; Homma, S.; Namiki, Y.; Ohkusa, T.; Gong, J.; Tajiri, H. Cancer vaccine by fusions of dendritic and cancer cells. Clin. Dev. Immunol. 2009, 2009, 657369. [Google Scholar] [CrossRef]
- Koido, S.; Hara, E.; Homma, S.; Torii, A.; Toyama, Y.; Kawahara, H.; Watanabe, M.; Yanaga, K.; Fujise, K.; Tajiri, H.; et al. Dendritic cells fused with allogeneic colorectal cancer cell line present multiple colorectal cancer-specific antigens and induce antitumor immunity against autologous tumor cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 7891–7900. [Google Scholar] [CrossRef] [Green Version]
- Koido, S.; Hara, E.; Homma, S.; Mitsunaga, M.; Takahara, A.; Nagasaki, E.; Kawahara, H.; Watanabe, M.; Toyama, Y.; Yanagisawa, S.; et al. Synergistic induction of antigen-specific CTL by fusions of TLR-stimulated dendritic cells and heat-stressed tumor cells. J. Immunol. 2007, 179, 4874–4883. [Google Scholar] [CrossRef] [Green Version]
- Krause, S.W.; Neumann, C.; Soruri, A.; Mayer, S.; Peters, J.H.; Andreesen, R. The treatment of patients with disseminated malignant melanoma by vaccination with autologous cell hybrids of tumor cells and dendritic cells. J. Immunother. 2002, 25, 421–428. [Google Scholar] [CrossRef]
- Haenssle, H.A.; Krause, S.W.; Emmert, S.; Zutt, M.; Kretschmer, L.; Schmidberger, H.; Andreesen, R.; Soruri, A. Hybrid cell vaccination in metastatic melanoma: Clinical and immunologic results of a phase I/II study. J. Immunother. 2004, 27, 147–155. [Google Scholar] [CrossRef]
- Pinho, M.P.; Sundarasetty, B.S.; Bergami-Santos, P.C.; Steponavicius-Cruz, K.; Ferreira, A.K.; Stripecke, R.; Barbuto, J.A. Dendritic-tumor cell hybrids induce tumor-specific immune responses more effectively than the simple mixture of dendritic and tumor cells. Cytotherapy 2016, 18, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Geskin, L.J.; Damiano, J.J.; Patrone, C.C.; Butterfield, L.H.; Kirkwood, J.M.; Falo, L.D. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res. 2018, 28, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Niedzwiecki, D.; Marshall, J.L.; Garrett, C.; Chang, D.Z.; Aklilu, M.; Crocenzi, T.S.; Cole, D.J.; Dessureault, S.; Hobeika, A.C.; et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann. Surg. 2013, 258, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Smed-Sorensen, A.; Cohn, L.; Chalouni, C.; Vandlen, R.; Lee, B.C.; Widger, J.; Keler, T.; Delamarre, L.; Mellman, I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012, 120, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Sittig, S.P.; Blom, R.A.; Cruz, L.J.; Schreibelt, G.; Figdor, C.G.; de Vries, I.J. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J. Immunol. 2013, 191, 5005–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillman, R.O.; McClay, E.F.; Barth, N.M.; Amatruda, T.T.; Schwartzberg, L.S.; Mahdavi, K.; de Leon, C.; Ellis, R.E.; DePriest, C. Dendritic versus tumor cell presentation of autologous tumor antigens for active specific immunotherapy in metastatic melanoma: Impact on long-term survival by extent of disease at the time of treatment. Cancer Biother. Radiopharm. 2015, 30, 187–194. [Google Scholar] [CrossRef]
- Weng, D.; Calderwood, S.K.; Gong, J. A novel heat shock protein 70-based vaccine prepared from DC-tumor fusion cells. Methods Mol. Biol. 2018, 1709, 359–369. [Google Scholar] [CrossRef]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef]
- Chu, Y.; Liu, Q.; Wei, J.; Liu, B. Personalized cancer neoantigen vaccines come of age. Theranostics 2018, 8, 4238–4246. [Google Scholar] [CrossRef]
- Baldin, A.V.; Grishina, A.N.; Korolev, D.O.; Kuznetsova, E.B.; Golovastova, M.O.; Kalpinskiy, A.S.; Alekseev, B.Y.; Kaprin, A.D.; Zinchenko, D.V.; Savvateeva, L.V.; et al. Autoantibody against arrestin-1 as a potential biomarker of renal cell carcinoma. Biochimie 2019, 157, 26–37. [Google Scholar] [CrossRef]
- Golovastova, M.O.; Tsoy, L.V.; Bocharnikova, A.V.; Korolev, D.O.; Gancharova, O.S.; Alekseeva, E.A.; Kuznetsova, E.B.; Savvateeva, L.V.; Skorikova, E.E.; Strelnikov, V.V.; et al. The cancer-retina antigen recoverin as a potential biomarker for renal tumors. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 9899–9907. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Human tumor antigens yesterday, today, and tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Riviere, I.; Sadelain, M. Chimeric antigen receptors: A cell and gene therapy perspective. Mol. Ther. 2017, 25, 1117–1124. [Google Scholar] [CrossRef]
- Christofi, T.; Baritaki, S.; Falzone, L.; Libra, M.; Zaravinos, A. Current perspectives in cancer immunotherapy. Cancers (Basel) 2019, 11, 1472. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| DC (APC) Targeting Method | Condition | Trial Phase | Trial ID | Refs |
|---|---|---|---|---|
| MR targeting | Different advanced cancers | I | NCT00709462; NCT00648102 | [44] |
| DC-SIGN targeting | Different NY-ESO-1-expressing tumors | I | NCT02122861 | [45] |
| DC-SIGN targeting | Melanoma | I | N/A | [46] |
| DEC-205 targeting | Different advanced cancers | I | NCT00948961 | [47] |
| SR targeting with HSP | Pancreatic adenocarcinoma | I | N/A | [48] |
| SR targeting with HSP | Glioblastoma | I | NCT02122822; ChiCTR-ONC-13003309 | [49] |
| SR targeting with HSP | Glioblastoma | II | NCT00905060 | [50] |
| SR targeting with HSP | Glioblastoma | II | NCT00293423 | [51] |
| SR targeting with HSP | Cervical intraepithelial neoplasia III | II | NCT00075569 | [52,53] |
| SR targeting with HSP | Melanoma | II | N/A | [54] |
| SLOs DCs targeting | Melanoma | II | N/A | [55] |
| DC Subset | DEC205/CD205 | MR/CD206 | Langerin/CD207 | DC-SIGN/CD209 | BDCA2/CD303 | Clec4A/DCIR | Clec9A/DNGR1 | Clec12A |
|---|---|---|---|---|---|---|---|---|
| moDC 1 | + | + | − | + | − | + | − | + |
| Blood-resident DCs | ||||||||
| cDC1 | + | − | − | − | − | + | + | + |
| cDC2 | + | − | − | − | − | + | − | + |
| CD16+ DC 2 | + | − | − | − | − | + | − | ? |
| pDC | + | − | − | − | + | + | − | + |
| Tissue-resident DCs | ||||||||
| cDC1 | ? | − | − | − | − | + | + | ? |
| cDC2 | − | ? 3 | ± 4 | − | − | ? | − | ? |
| LCs 5 | + | − | + | − | − | ? | ? | ? |
| IDECs | ? | + 6 | − | +6 | ? | ? | ? | ? |
| Type of Study | Type of Vaccine | Combinatorial Treatment | Detected Response | Refs | |
|---|---|---|---|---|---|
| DC Target/Source | Targeting/Loading Method | ||||
| In vitro | cDC2 or moDCs ex vivo targeting | Anti-DEC205 targeting | LPS | CD4+ and CD8+ T cells | [254] |
| Anti-MR targeting | |||||
| Anti-CD40 targeting | |||||
| In vitro | pDC ex vivo targeting | Anti-DEC205 NPs | Each NP encapsulate R848 TLR7 agonist | CD4+ and CD8+ T cells | [255] |
| Anti-DCIR NPs | |||||
| Anti-BDCA2 NPs | |||||
| Anti- FcγRIIa NPs | |||||
| In vitro | moDCs ex vivo loading | Coculture of moDCs with MDA-MB-231 or KA2 cells | CD8+ T cells | [251] | |
| moDCs ex vivo fusion | Electroporation-mediated fusion of moDCs with MDA-MB-231 or KA2 cells | CD8+ T cells | |||
| Phase II clinical trial | In vivo targeting | Administration of poxvector | GM-CSF | T-cell response | [253] |
| moDCs ex vivo loading | Loading with poxvector | ||||
| Phase II clinical trial | In vivo targeting | Administration of autologous tumor-derived cells | N/A | [256] | |
| moDCs ex vivo loading | Coculture with autologous tumor-derived cells | ||||
| Phase II clinical trial | moDCs ex vivo loading | Coculture with autologous tumor cells | CD8+ T cells; DTH-test | [252] | |
| moDCs ex vivo fusion | PEG-mediated fusion with autologous tumor cells | CD8+ T cells; DTH-test | |||
| moDCs ex vivo loading | Pulsing with autologous tumor cell lysate | CD8+ T cells; DTH-test | |||
| Pros/Cons | In Vivo Targeting | Ex Vivo Loading |
|---|---|---|
| Pros | Off the shelf use:
| Highly controlled maturation and activation:
|
| Cons | Poor control of maturation and activation:
| Tailor made:
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A., Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. https://doi.org/10.3390/cancers12030590
Baldin AV, Savvateeva LV, Bazhin AV, Zamyatnin AA Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers. 2020; 12(3):590. https://doi.org/10.3390/cancers12030590
Chicago/Turabian StyleBaldin, Alexey V., Lyudmila V. Savvateeva, Alexandr V. Bazhin, and Andrey A. Zamyatnin, Jr. 2020. "Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting" Cancers 12, no. 3: 590. https://doi.org/10.3390/cancers12030590