Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor

 , , , , , ,
, , , , , , 
Abstract
:1. Introduction
2. Results
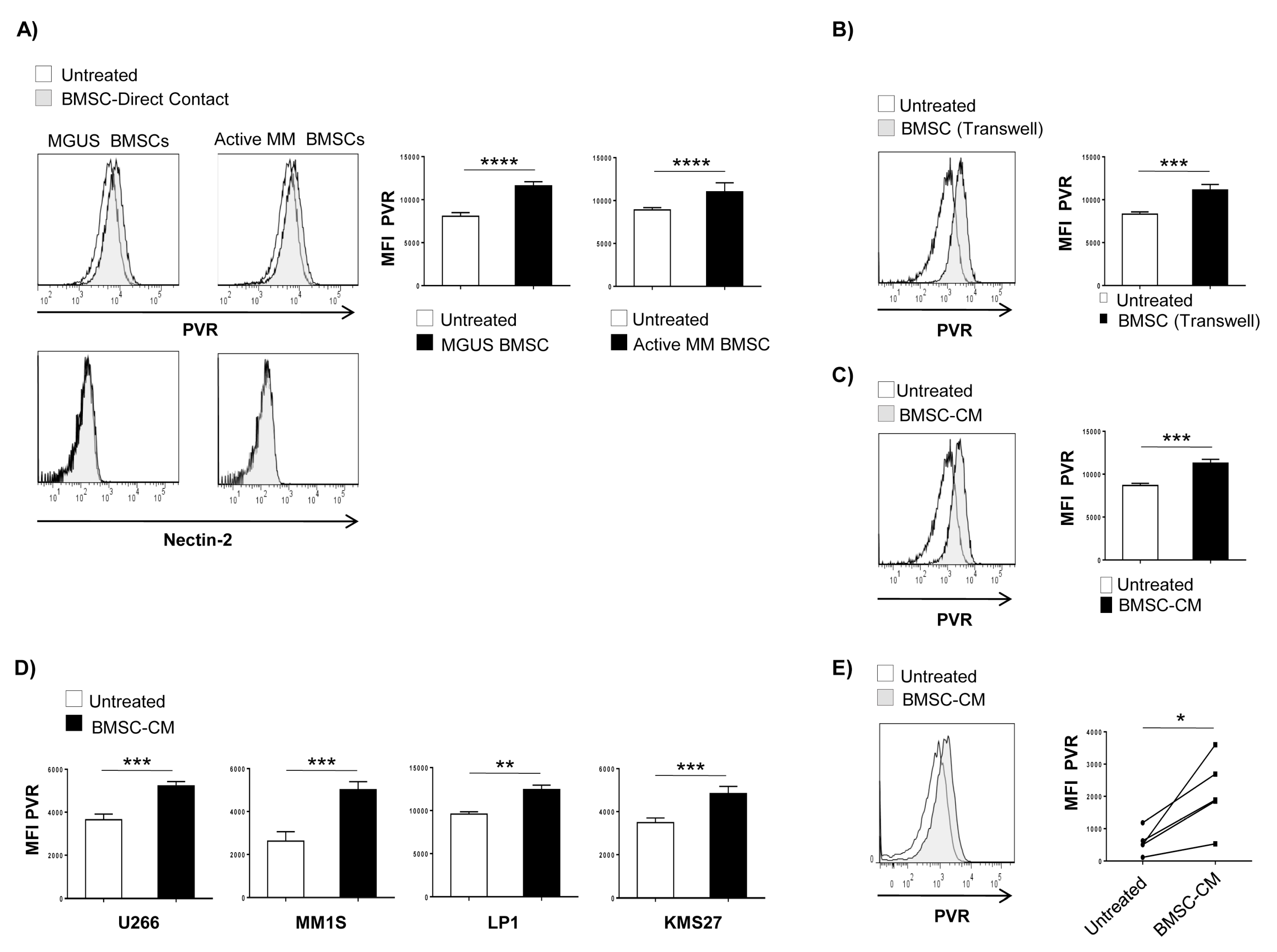
2.1. BMSC-Derived Soluble Factor(s) Increase PVR Surface Expression on MM Cells
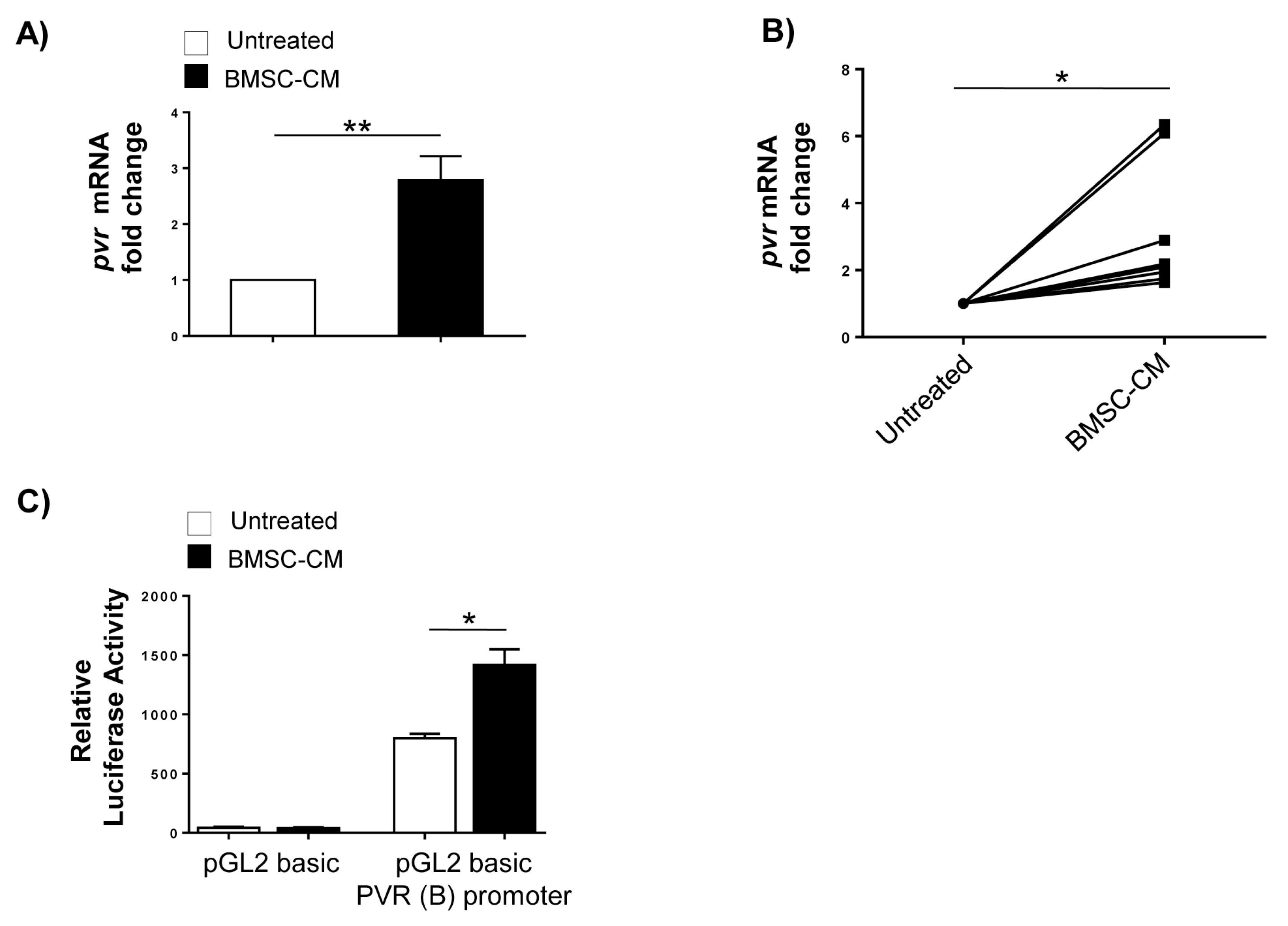
2.2. Transcriptional Control of PVR Expression on MM Cells by BMSCs: Role of the Transcription Factor NF-kB
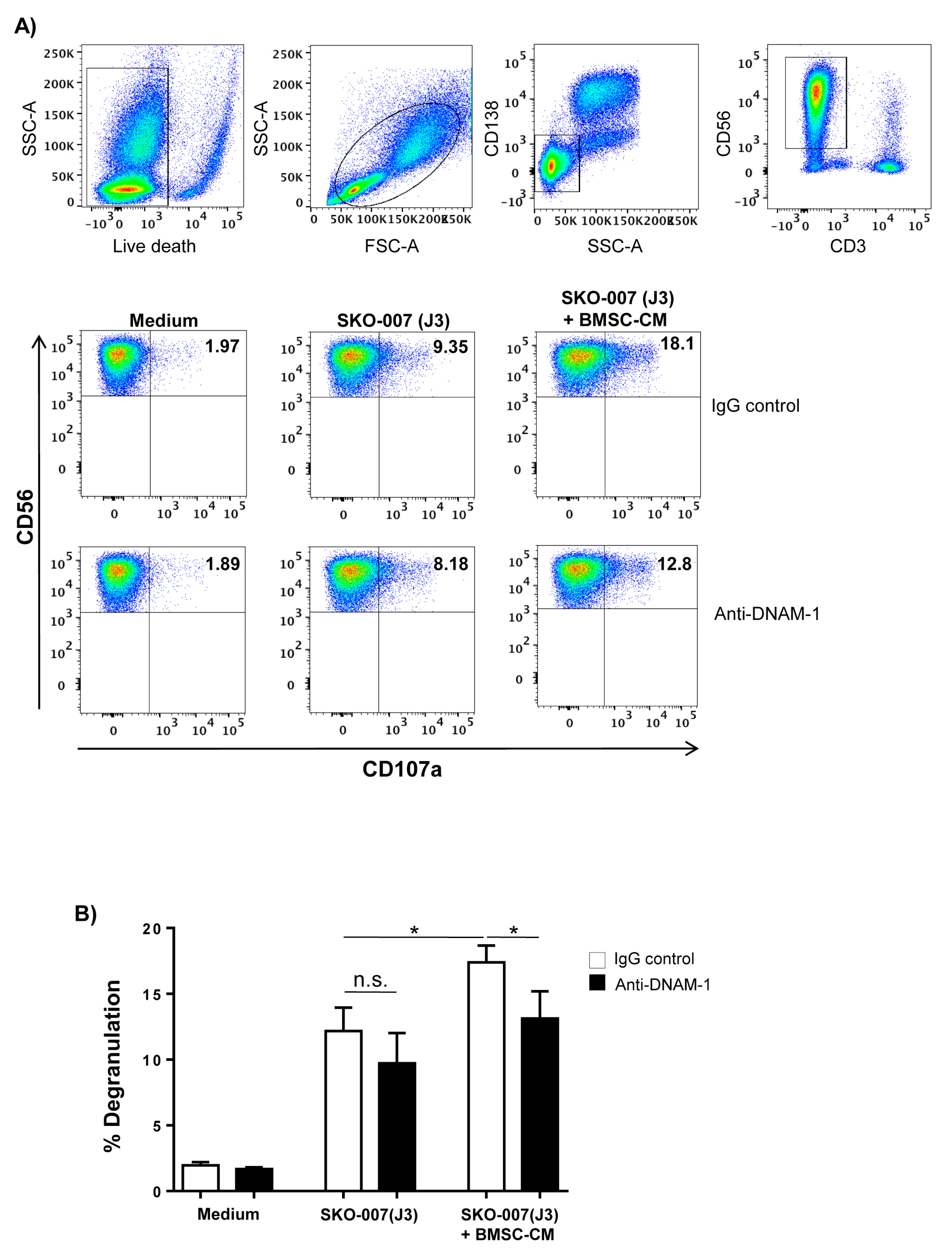
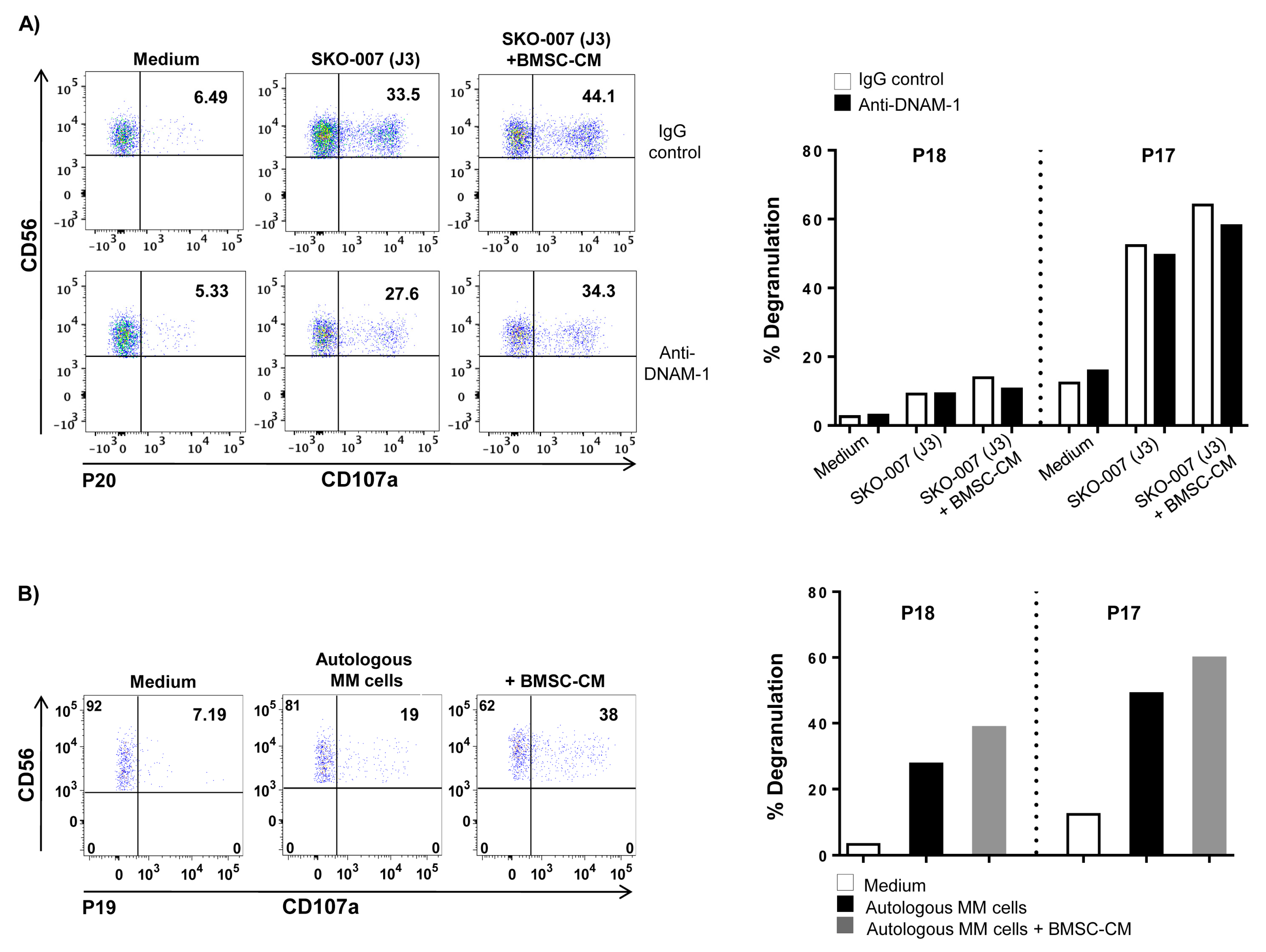
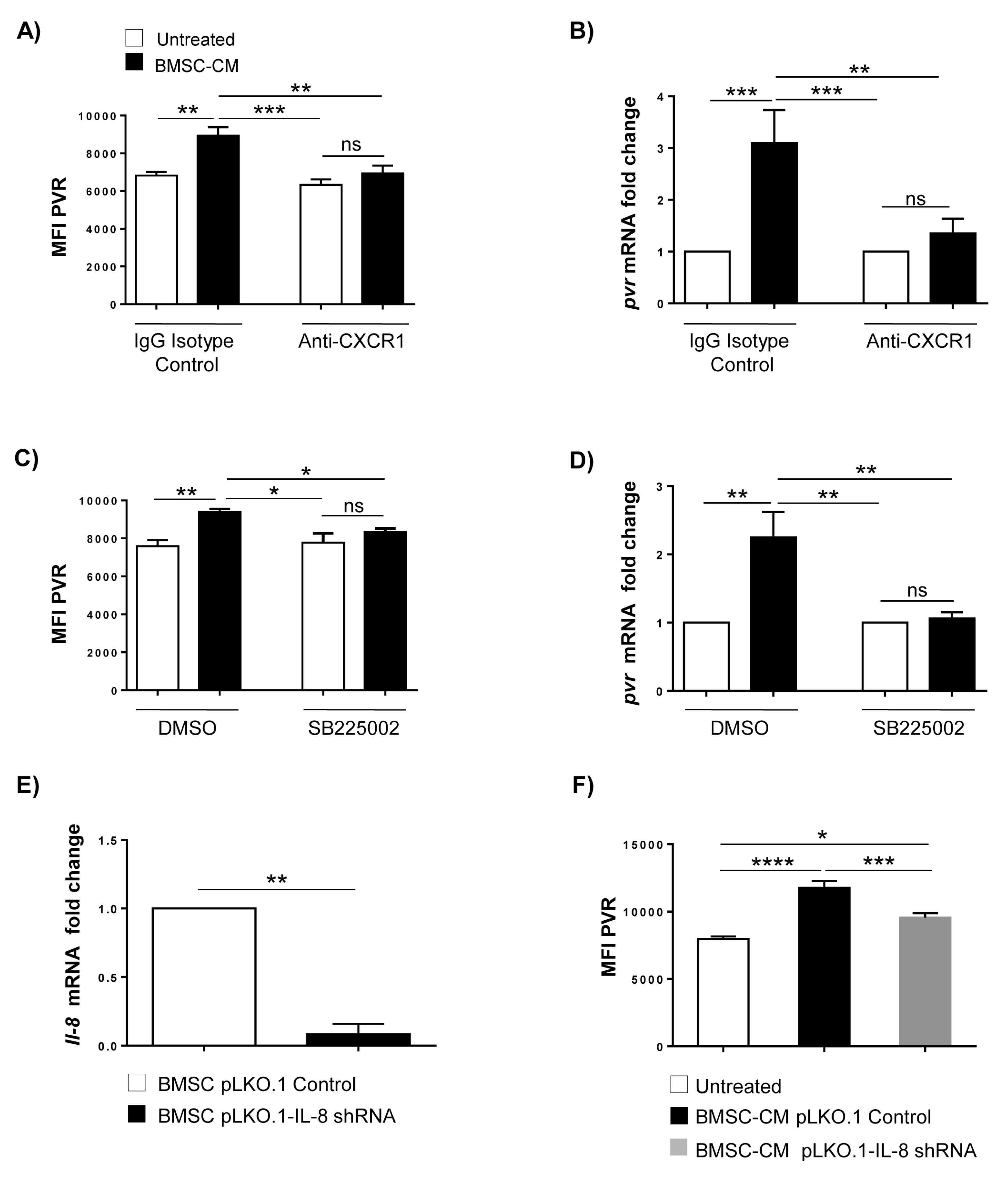
2.3. Increased PVR Expression on MM Cells by BMSCs Involves IL-8
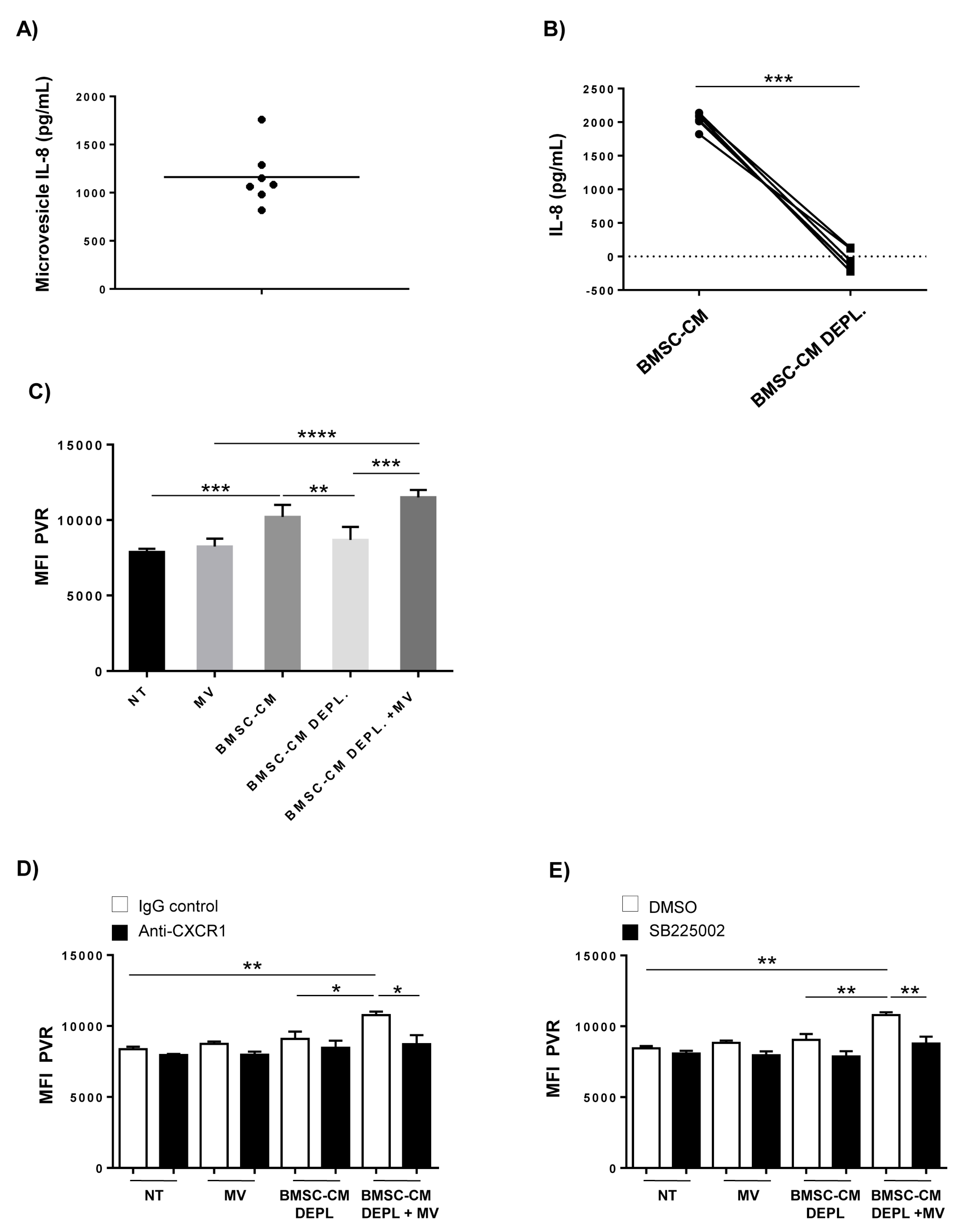
2.4. Requirement of IL-8-Bearing Microvesicles for BMSC-Induced PVR Upregulation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Clinical Samples
4.2. Adipogenic and Osteogenic Differentiation
4.3. Reagents and Antibodies
4.4. Flow Cytometry and Degranulation Assay
4.5. Western-Blot Analysis
4.6. Plasmids
4.7. DNA Transfections, Virus Production and In Vitro Transduction
4.8. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.9. Annexin V
4.10. Enzyme-Linked Immunosorbent Assay (ELISA)
4.11. Preparation of BMSC-CM and Microvesicles
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kawano, Y.; Roccaro, A.M.; Ghobrial, I.M.; Azzi, J. Multiple Myeloma and the Immune Microenvironment. Curr. Cancer Drug Targets 2017, 17, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Dosani, T.; Mailankody, S.; Korde, N.; Manasanch, E.; Bhutani, M.; Tageja, N.; Roschewski, M.; Kwok, M.; Kazandjian, D.; Costello, R.; et al. Host-related immunodeficiency in the development of multiple myeloma. Leuk. Lymphoma 2018, 59, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Podar, K.; Breitkreutz, I.; Richardson, P.G.; Anderson, K.C. Multiple myeloma. Lancet 2009, 374, 324–339. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gomez, A.; de Las, R.J.; Ocio, E.M.; Diaz-Rodriguez, E.; Montero, J.C.; Martin, M.; Blanco, J.F.; Sanchez-Guijo, F.M.; Pandiella, A.; Miguel, J.F.S.; et al. Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: Implications in myeloma progression and myeloma bone disease. Oncotarget 2014, 5, 8284–8305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, M.; Ohkawara, H.; Ogawa, K.; Ikeda, K.; Ueda, K.; Shichishima-Nakamura, A.; Ito, E.; Imai, J.I.; Yanagisawa, Y.; Honma, R.; et al. Autocrine and Paracrine Interactions between Multiple Myeloma Cells and Bone Marrow Stromal Cells by Growth Arrest-specific Gene 6 Cross-talk with Interleukin-6. J. Biol. Chem. 2017, 292, 4280–4292. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Chauhan, D.; Hayashi, T.; Podar, K.; Akiyama, M.; Gupta, D.; Richardson, P.; Munshi, N.; Anderson, K.C. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol. Cancer Ther. 2002, 1, 539–544. [Google Scholar]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Matthews, G.M.; de Matos, S.R.; Dhimolea, E.; Sheffer, M.; Gandolfi, S.; Dashevsky, O.; Sorrell, J.D.; Mitsiades, C.S. NF-κB dysregulation in multiple myeloma. Semin. Cancer Biol. 2016, 39, 68–76. [Google Scholar] [CrossRef]
- Bharti, A.C.; Shishodia, S.; Reuben, J.M.; Weber, D.; Alexanian, R.; Raj-Vadhan, S.; Estrov, Z.; Talpaz, M.; Aggarwal, B.B. Nuclear factor-κB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004, 103, 3175–3184. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De, B.E.; Van, V.E.; Lahoutte, T.; De, W.O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Faict, S.; Maes, K.; De, B.E.; Van, V.E.; Schots, R.; Vanderkerken, K.; Menu, E. Extracellular vesicle cross-talk in the bone marrow microenvironment: Implications in multiple myeloma. Oncotarget 2016, 7, 38927–38945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, H.; Attar-Schneider, O.; Dabbah, M.; Zismanov, V.; Tartakover-Matalon, S.; Lishner, M.; Drucker, L. Mesenchymal stem cells secretomes’ affect multiple myeloma translation initiation. Cell Signal. 2016, 28, 620–630. [Google Scholar] [CrossRef]
- Dabbah, M.; Attar-Schneider, O.; Tartakover, M.S.; Shefler, I.; Jarchwsky, D.O.; Lishner, M.; Drucker, L. Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells’ phenotype and translation initiation. Carcinogenesis 2017, 38, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Garayoa, M.; Garcia, J.L.; Santamaria, C.; Garcia-Gomez, A.; Blanco, J.F.; Pandiella, A.; Hernandez, J.M.; Sanchez-Guijo, F.M.; del Canizo, M.C.; Gutierrez, N.C.; et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia 2009, 23, 1515–1527. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Sanchez-Guijo, F.; del Canizo, M.C.; Miguel, J.F.S.; Garayoa, M. Multiple myeloma mesenchymal stromal cells: Contribution to myeloma bone disease and therapeutics. World J. Stem Cells 2014, 6, 322–343. [Google Scholar] [CrossRef]
- Wallace, S.R.; Oken, M.M.; Lunetta, K.L.; Panoskaltsis-Mortari, A.; Masellis, A.M. Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer 2001, 91, 1219–1230. [Google Scholar] [CrossRef]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; de, A.L.F.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Investig. 2015, 125, 2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di, G.; Cippitelli, V.M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M.G. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsten, M.; Bjorkstrom, N.K.; Norell, H.; Bryceson, Y.; van, H.T.; Baumann, B.C.; Hanson, M.; Schedvins, K.; Kiessling, R.; Ljunggren, H.G.; et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007, 67, 1317–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Stabile, H.; Molfetta, R.; Soriani, A.; Bernardini, G.; Zingoni, A.; Gismondi, A.; Paolini, R.; Cippitelli, M.; Santoni, A. Translating the anti-myeloma activity of Natural Killer cells into clinical application. Cancer Treat. Rev. 2018, 70, 255–264. [Google Scholar] [CrossRef]
- Fionda, C.; Soriani, A.; Zingoni, A.; Santoni, A.; Cippitelli, M. NKG2D and DNAM-1 Ligands: Molecular Targets for NK Cell-Mediated Immunotherapeutic Intervention in Multiple Myeloma. Biomed. Res. Int. 2015, 2015, 178698. [Google Scholar] [CrossRef]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Soriani, A.; Ricci, B.; Molfetta, R.; Paolini, R.; Santoni, A.; Cippitelli, M. Nitric oxide donors increase PVR/CD155 DNAM-1 ligand expression in multiple myeloma cells: Role of DNA damage response activation. BMC Cancer 2015, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Iannitto, M.L.; Ricci, B.; Fionda, C.; Malgarini, G.; Morrone, S.; Peruzzi, G.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J. Immunol. 2014, 193, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Borrelli, C.; Ricci, B.; Molfetta, R.; Zingoni, A.; Fionda, C.; Carnevale, S.; Abruzzese, M.P.; Petrucci, M.T.; Ricciardi, M.R.; et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology 2017, 6, e1264564. [Google Scholar] [CrossRef] [Green Version]
- Zitti, B.; Molfetta, R.; Fionda, C.; Quatrini, L.; Stabile, H.; Lecce, M.; de Turris, V.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Sci. Rep. 2017, 7, 10445. [Google Scholar] [CrossRef]
- Molfetta, R.; Milito, N.D.; Zitti, B.; Lecce, M.; Fionda, C.; Cippitelli, M.; Santoni, A.; Paolini, R. The Ubiquitin-proteasome pathway regulates Nectin2/CD112 expression and impairs NK cell recognition and killing. Eur. J. Immunol. 2019, 49, 873–883. [Google Scholar] [CrossRef]
- Dominici, M.; Le, B.K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L. Immunological characterization of multipotent mesenchymal stromal cells—The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.P.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Markovina, S.; Callander, N.S.; O’Connor, S.L.; Kim, J.; Werndli, J.E.; Raschko, M.; Leith, C.P.; Kahl, B.S.; Kim, K.; Miyamoto, S. Bortezomib-resistant nuclear factor-κB activity in multiple myeloma cells. Mol. Cancer Res. 2008, 6, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Huynh, M.; Pak, C.; Markovina, S.; Callander, N.S.; Chng, K.S.; Wuerzberger-Davis, S.M.; Bakshi, D.D.; Kink, J.A.; Hematti, P.; Hope, C.; et al. Hyaluronan and proteoglycan link protein 1 (HAPLN1) activates bortezomib-resistant NF-κB activity and increases drug resistance in multiple myeloma. J. Biol. Chem. 2018, 293, 2452–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsiades, C.S.; Mitsiades, N.S.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: Interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer 2006, 42, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Selitrennik, M.; Lev, S. PYK2 integrates growth factor and cytokine receptors signaling and potentiates breast cancer invasion via a positive feedback loop. Oncotarget 2015, 6, 22214–22226. [Google Scholar] [CrossRef] [Green Version]
- Petreaca, M.L.; Yao, M.; Liu, Y.; Defea, K.; Martins-Green, M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol. Biol. Cell 2007, 18, 5014–5023. [Google Scholar] [CrossRef] [Green Version]
- Kyriakakis, E.; Cavallari, M.; Pfaff, D.; Fabbro, D.; Mestan, J.; Philippova, M.; De, L.G.; Erne, P.; Resink, T.J. IL-8-mediated angiogenic responses of endothelial cells to lipid antigen activation of iNKT cells depend on EGFR transactivation. J. Leukoc. Biol. 2011, 90, 929–939. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Trieu, K.; Zhao, M.; Rose, D.M.; Terkeltaub, R.A.; Burger, M. IL-8-mediated cell migration in endothelial cells depends on cathepsin B activity and transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6714–6722. [Google Scholar] [CrossRef] [Green Version]
- Del, P.A.; Martinez-Munoz, L.; Mazzon, C.; Toffali, L.; Sozio, F.; Za, L.; Bosisio, D.; Gazzurelli, L.; Salvi, V.; Tiberio, L.; et al. The atypical receptor CCRL2 is required for CXCR2-dependent neutrophil recruitment and tissue damage. Blood 2017, 130, 1223–1234. [Google Scholar]
- Vulpis, E.; Stabile, H.; Soriani, A.; Fionda, C.; Petrucci, M.T.; Mariggio, E.; Ricciardi, M.R.; Cippitelli, M.; Gismondi, A.; Santoni, A.; et al. Key Role of the CD56(low)CD16(low) Natural Killer Cell Subset in the Recognition and Killing of Multiple Myeloma Cells. Cancers 2018, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Nakai, R.; Maniwa, Y.; Tanaka, Y.; Nishio, W.; Yoshimura, M.; Okita, Y.; Ohbayashi, C.; Satoh, N.; Ogita, H.; Takai, Y.; et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Sci. 2010, 101, 1326–1330. [Google Scholar] [CrossRef]
- Bevelacqua, V.; Bevelacqua, Y.; Candido, S.; Skarmoutsou, E.; Amoroso, A.; Guarneri, C.; Strazzanti, A.; Gangemi, P.; Mazzarino, M.C.; D’Amico, F.; et al. Nectin like-5 overexpression correlates with the malignant phenotype in cutaneous melanoma. Oncotarget 2012, 3, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A multi-functional molecule in tumor progression. Int. J. Mol. Sci. 2020, 21, 922. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Malgarini, G.; Soriani, A.; Zingoni, A.; Cecere, F.; Iannitto, M.L.; Ricciardi, M.R.; Federico, V.; Petrucci, M.T.; Santoni, A.; et al. Inhibition of glycogen synthase kinase-3 increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of STAT3. J. Immunol. 2013, 190, 6662–6672. [Google Scholar] [CrossRef] [PubMed]
- Capuano, C.; Pighi, C.; Molfetta, R.; Paolini, R.; Battella, S.; Palmieri, G.; Giannini, G.; Belardinilli, F.; Santoni, A.; Galandrini, R. Obinutuzumab-mediated high-affinity ligation of FcgammaRIIIA/CD16 primes NK cells for IFNgamma production. Oncoimmunology 2017, 6, e1290037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabile, H.; Carlino, C.; Mazza, C.; Giliani, S.; Morrone, S.; Notarangelo, L.D.; Notarangelo, L.D.; Santoni, A.; Gismondi, A. Impaired NK-cell migration in WAS/XLT patients: Role of Cdc42/WASp pathway in the control of chemokine-induced beta2 integrin high-affinity state. Blood 2010, 115, 2818–2826. [Google Scholar] [CrossRef] [Green Version]
- Cippitelli, M.; Fionda, C.; Di, B.D.; Di, R.F.; Lupo, A.; Piccoli, M.; Frati, L.; Santoni, A. Negative regulation of CD95 ligand gene expression by vitamin D3 in T lymphocytes. J. Immunol. 2002, 168, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Cippitelli, M.; Fionda, C.; Di, B.D.; Lupo, A.; Piccoli, M.; Frati, L.; Santoni, A. The cyclopentenone-type prostaglandin 15-deoxy-delta 12,14-prostaglandin J2 inhibits CD95 ligand gene expression in T lymphocytes: Interference with promoter activation via peroxisome proliferator-activated receptor-gamma-independent mechanisms. J. Immunol. 2003, 170, 4578–4592. [Google Scholar] [CrossRef] [Green Version]
- Solecki, D.; Wimmer, E.; Lipp, M.; Bernhardt, G. Identification and characterization of the cis-acting elements of the human CD155 gene core promoter. J. Biol. Chem. 1999, 274, 1791–1800. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Soriani, A.; Malgarini, G.; Iannitto, M.L.; Santoni, A.; Cippitelli, M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J. Immunol. 2009, 183, 4385–4394. [Google Scholar] [CrossRef] [Green Version]
- Vulpis, E.; Cecere, F.; Molfetta, R.; Soriani, A.; Fionda, C.; Peruzzi, G.; Caracciolo, G.; Palchetti, S.; Masuelli, L.; Simonelli, L.; et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology 2017, 6, e1279372. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex/Age | Clinical Stage | Monoclonal Ig | % of PCs |
|---|---|---|---|---|
| 1 | M/74 | MM PD | IgG-ʎ | 8 |
| 2 | F/63 | Relapse | IgGλ | 90 |
| 3 | M/65 | Relapse | IgG-ʎ | 70 |
| 4 | M/61 | Relapse | IgG-k | 55 |
| 5 | M/67 | Relapse | IgG-k | 15 |
| 6 | M/73 | Onset | IgG-ʎ | 61 |
| 7 | F/74 | Onset | IgG-ʎ | 23 |
| 8 | M/65 | Relapse | IgG-ʎ | 29 |
| 9 | F/74 | Smoldering | IgG-k | 64 |
| 10 | F/70 | Onset | IgG-k | 48 |
| 11 | M/79 | Relapse | IgG-k | 34 |
| 12 | M/59 | Smoldering | IgGk | 40 |
| 13 | M/55 | Onset | IgG-k | 38 |
| 14 | M/61 | Relapse | IgG-k | 57 |
| 15 | F/57 | Smoldering | IgG-ʎ | 10 |
| 16 | F/75 | MGUS | IgG-ʎ | 7 |
| 17 | M/80 | Onset | IgG-k | 25 |
| 18 | M/88 | Onset | IgG-k | 15 |
| 19 | F/60 | Onset | IgG-ʎ | 25 |
| 20 | F/71 | Smoldering | IgG-k | 4 |
| Patient | Sex/Age | Clinical Stage | Monoclonal Ig | % of PCs |
|---|---|---|---|---|
| 1 | M/74 | MM PD | IgG-ʎ | 8 |
| 2 | F/63 | Relapse | IgGλ | 90 |
| 3 | M/65 | Relapse | IgG-ʎ | 70 |
| 4 | F/76 | Onset | IgG-k | 55 |
| 5 | M/67 | Relapse | IgG-k | 15 |
| 6 | M/61 | Onset | IgG-ʎ | 61 |
| 7 | F/74 | Onset | IgG-ʎ | 23 |
| 8 | M/73 | Relapse | IgG-ʎ | 29 |
| 9 | F/74 | Smoldering | IgG-ʎ | 64 |
| 10 | F/70 | Onset | IgG-k | 48 |
| 11 | F/62 | Onset | IgG-k | 48 |
| 12 | M/59 | Smoldering | IgG-k | 69 |
| 13 | M/68 | Relapse | IgG-k | 9 |
| 14 | F/65 | Onset | IgG-ʎ | 73 |
| 15 | F/67 | Onset | IgG-ʎ | 22 |
| 16 | M/79 | Relapse | IgG-k | 34 |
| 17 | M/55 | Onset | IgG-k | 38 |
| 18 | M/61 | Relapse | IgG-k | 57 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekhloufi, A.; Kosta, A.; Stabile, H.; Molfetta, R.; Zingoni, A.; Soriani, A.; Cippitelli, M.; Paolini, R.; Gismondi, A.; Ricciardi, M.R.; et al. Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor. Cancers 2020, 12, 440. https://doi.org/10.3390/cancers12020440
Mekhloufi A, Kosta A, Stabile H, Molfetta R, Zingoni A, Soriani A, Cippitelli M, Paolini R, Gismondi A, Ricciardi MR, et al. Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor. Cancers. 2020; 12(2):440. https://doi.org/10.3390/cancers12020440
Chicago/Turabian StyleMekhloufi, Abdelilah, Andrea Kosta, Helena Stabile, Rosa Molfetta, Alessandra Zingoni, Alessandra Soriani, Marco Cippitelli, Rossella Paolini, Angela Gismondi, Maria Rosaria Ricciardi, and et al. 2020. "Bone Marrow Stromal Cell-Derived IL-8 Upregulates PVR Expression on Multiple Myeloma Cells via NF-kB Transcription Factor" Cancers 12, no. 2: 440. https://doi.org/10.3390/cancers12020440