EVI1 as a Prognostic and Predictive Biomarker of Clear Cell Renal Cell Carcinoma

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. EVI1 Overexpression Is Associated with Progression Features and Poor Prognosis of ccRCC
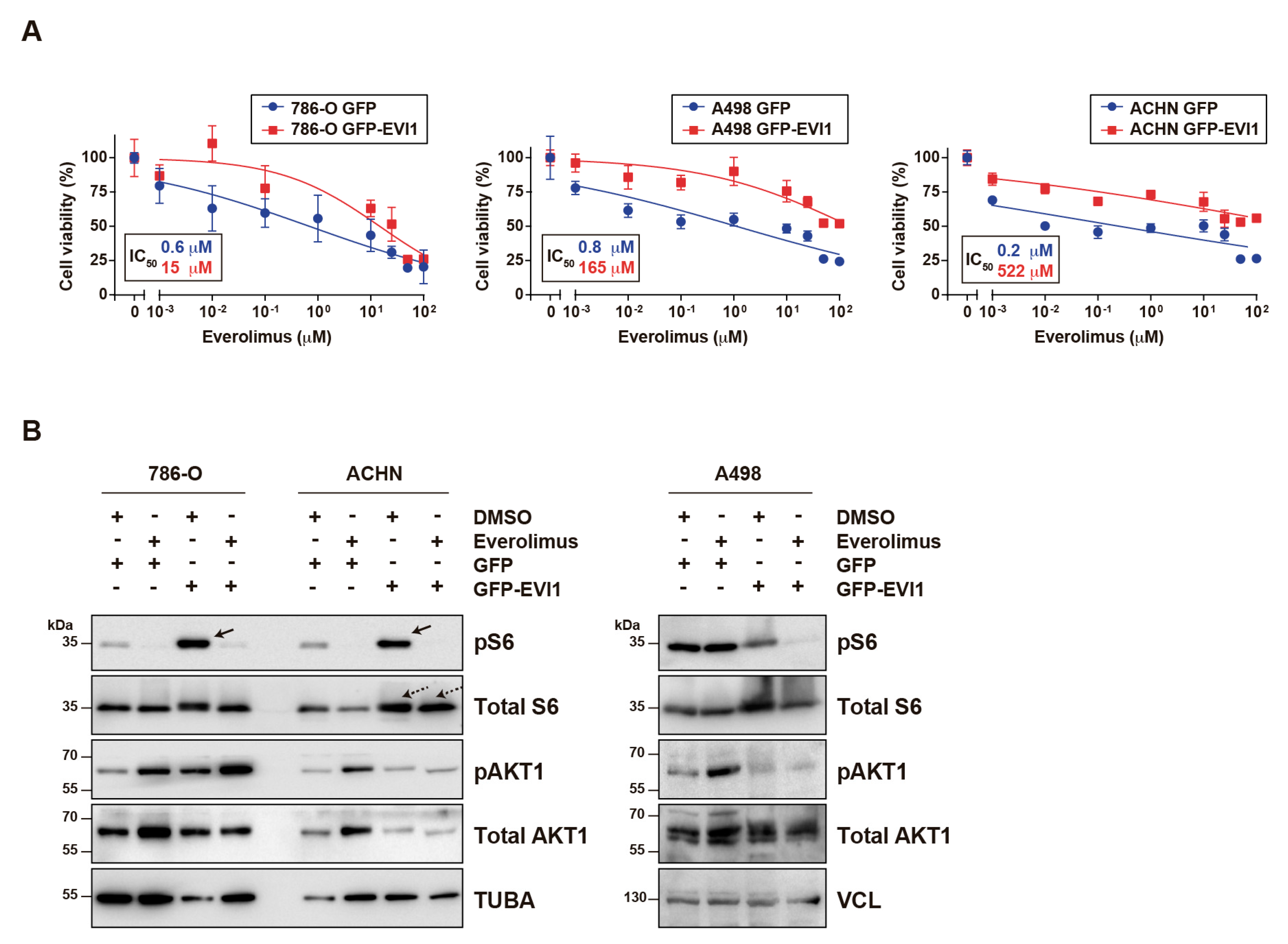
2.2. EVI1 Overexpression Confers ccRCC Cell Resistance to Everolimus
2.3. Common Genetic Variants in EVI1 Are Associated with Response to Everolimus of Metastatic ccRCC
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Immunohistochemistry Assays
4.3. Genetic Analyses
4.4. Statistical Analyses
4.5. TCGA Data Analyses
4.6. Cellular Assays
4.7. Western Blot and Antibodies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mucenski, M.L.; Taylor, B.A.; Ihle, J.N.; Hartley, J.W.; Morse, H.C.; Jenkins, N.A.; Copeland, N.G. Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol. Cell. Biol. 1988, 8, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Kurokawa, M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci. 2012, 103, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Barjesteh van Waalwijk van Doorn-Khosrovani, S.; Erpelinck, C.; van Putten, W.L.J.; Valk, P.J.M.; van der Poel-van de Luytgaarde, S.; Hack, R.; Slater, R.; Smit, E.M.E.; Beverloo, H.B.; Verhoef, G.; et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood 2003, 101, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Lugthart, S.; Hollink, I.H.; Arentsen-Peters, S.T.J.C.M.; van Wering, E.R.; de Graaf, S.S.N.; Reinhardt, D.; Creutzig, U.; Kaspers, G.J.L.; de Bont, E.S.J.M.; et al. EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 2010, 24, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayadi, A.; Jeyakani, J.; Seet, S.H.; Wei, C.-L.; Bourque, G.; Bard, F.A.; Jenkins, N.A.; Copeland, N.G.; Bard-Chapeau, E.A. Functional features of EVI1 and EVI1Δ324 isoforms of MECOM gene in genome-wide transcription regulation and oncogenicity. Oncogene 2016, 35, 2311–2321. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Ko, T.C.; Fields, A.P.; Thompson, E.A. Evi1 is a survival factor which conveys resistance to both TGFb- and taxol-mediated cell death via PI3K/AKT. Oncogene 2006, 25, 3565–3575. [Google Scholar] [CrossRef] [Green Version]
- Mateo, F.; Arenas, E.J.; Aguilar, H.; Serra-Musach, J.; de Garibay, G.R.; Boni, J.; Maicas, M.; Du, S.; Iorio, F.; Herranz-Ors, C.; et al. Stem cell-like transcriptional reprogramming mediates metastatic resistance to mTOR inhibition. Oncogene 2017, 36, 2737–2749. [Google Scholar] [CrossRef]
- Wang, H.; Schaefer, T.; Konantz, M.; Braun, M.; Varga, Z.; Paczulla, A.M.; Reich, S.; Jacob, F.; Perner, S.; Moch, H.; et al. Prominent oncogenic roles of EVI1 in breast carcinoma. Cancer Res. 2017, 77, 2148–2160. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Suzuki, H.I.; Shibahara, J.; Kunita, A.; Isagawa, T.; Yoshimi, A.; Kurokawa, M.; Miyazono, K.; Aburatani, H.; Ishikawa, S.; et al. EVI1 oncogene promotes KRAS pathway through suppression of microRNA-96 in pancreatic carcinogenesis. Oncogene 2014, 33, 2454–2463. [Google Scholar] [CrossRef]
- Queisser, A.; Hagedorn, S.; Wang, H.; Schaefer, T.; Konantz, M.; Alavi, S.; Deng, M.; Vogel, W.; von Mässenhausen, A.; Kristiansen, G.; et al. Ecotropic viral integration site 1, a novel oncogene in prostate cancer. Oncogene 2017, 36, 1573–1584. [Google Scholar] [CrossRef]
- Choi, Y.-W.; Choi, J.S.; Zheng, L.T.; Lim, Y.J.; Yoon, H.K.; Kim, Y.H.; Wang, Y.-P.; Lim, Y. Comparative genomic hybridization array analysis and real time PCR reveals genomic alterations in squamous cell carcinomas of the lung. Lung Cancer. 2007, 55, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.T.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Wu, L.; Li, X.; Liu, X.; Ma, P.; Juang, Y. Ecotropic virus integration-1 and calreticulin as novel prognostic markers in triple-negative breast cancer: A retrospective cohort study. Oncol. Lett. 2019, 18, 1847–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanjundan, M.; Nakayama, Y.; Cheng, K.W.; Lahad, J.; Liu, J.; Lu, K.; Kuo, W.-L.; Smith-McCune, K.; Fishman, D.; Gray, J.W.; et al. Amplification of MDS1/EVI1 and EVI1, located in the 3q26.2 amplicon, is associated with favorable patient prognosis in ovarian cancer. Cancer Res. 2007, 67, 3074–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Bui, T.; Bauckman, K.A.; Keyomarsi, K.; Mills, G.B.; Nanjundan, M. EVI1 splice variants modulate functional responses in ovarian cancer cells. Mol. Oncol. 2013, 7, 647–668. [Google Scholar] [CrossRef]
- Bard-Chapeau, E.A.; Gunaratne, J.; Kumar, P.; Chua, B.Q.; Muller, J.; Bard, F.A.; Blackstock, W.; Copeland, N.G.; Jenkins, N.A. EVI1 oncoprotein interacts with a large and complex network of proteins and integrates signals through protein phosphorylation. Proc. Natl. Acad. Sci. USA 2013, 110, E2885–E2894. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Kurokawa, M. Evi1 forms a bridge between the epigenetic machinery and signaling pathways. Oncotarget 2011, 2, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Paredes, R.; Schneider, M.; Stevens, A.; White, D.J.; Williamson, A.J.K.; Muter, J.; Pearson, S.; Kelly, J.R.; Connors, K.; Wiseman, D.H.; et al. EVI1 carboxy-terminal phosphorylation is ATM-mediated and sustains transcriptional modulation and self-renewal via enhanced CtBP1 association. Nucleic Acids Res. 2018, 46, 7662–7674. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Goyama, S.; Watanabe-Okochi, N.; Yoshiki, Y.; Nannya, Y.; Nitta, E.; Arai, S.; Sato, T.; Shimabe, M.; Nakagawa, M.; et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 2011, 117, 3617–3628. [Google Scholar] [CrossRef] [Green Version]
- Lugthart, S.; Figueroa, M.E.; Bindels, E.; Skrabanek, L.; Valk, P.J.M.; Li, Y.; Meyer, S.; Erpelinck-Verschueren, C.; Greally, J.; Löwenberg, B.; et al. Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1. Blood 2011, 117, 234–241. [Google Scholar] [CrossRef]
- Cattaneo, F.; Nucifora, G. EVI1 recruits the histone methyltransferase SUV39H1 for transcription repression. J. Cell. Biochem. 2008, 105, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Goyama, S.; Nitta, E.; Yoshino, T.; Kako, S.; Watanabe-Okochi, N.; Shimabe, M.; Imai, Y.; Takahashi, K.; Kurokawa, M. EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia 2010, 24, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spensberger, D.; Delwel, R. A novel interaction between the proto-oncogene Evi1 and histone methyltransferases, SUV39H1 and G9a. FEBS Lett. 2008, 582, 2761–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, M.; Mitani, K.; Irie, K.; Matsuyama, T.; Takahashi, T.; Chiba, S.; Yazaki, Y.; Matsumoto, K.; Hirai, H. The oncoprotein Evi-1 represses TGF-b signalling by inhibiting Smad3. Nature 1998, 394, 92–96. [Google Scholar] [CrossRef]
- Izutsu, K.; Kurokawa, M.; Imai, Y.; Maki, K.; Mitani, K.; Hirai, H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood 2001, 97, 2815–2822. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Keefe, S.M.; Nathanson, K.L.; Rathmell, W.K. The molecular biology of renal cell carcinoma. Semin. Oncol. 2013, 40, 421–428. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Wagner, B.; Patard, J.-J.; Méjean, A.; Bensalah, K.; Verhoest, G.; Zigeuner, R.; Ficarra, V.; Tostain, J.; Mulders, P.; Chautard, D.; et al. Prognostic value of renal vein and inferior vena cava involvement in renal cell carcinoma. Eur. Urol. 2009, 55, 452–459. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Molinie, V.; Bracarda, S.; Maroto, P.; Szczylik, C.; Nathan, P.; Negrier, S.; Weiss, C.; Porta, C.; Grünwald, V.; et al. Open-label phase 2 trial of first-line everolimus monotherapy in patients with papillary metastatic renal cell carcinoma: RAPTOR final analysis. Eur. J. Cancer. 1990 2016, 69, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler Artigas, M.; Loth, D.W.; Wain, L.V.; Gharib, S.A.; Obeidat, M.; Tang, W.; Zhai, G.; Zhao, J.H.; Smith, A.V.; Huffman, J.E.; et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 2011, 43, 1082–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Sim, X.; Go, M.J.; Wu, J.-Y.; Gu, D.; Takeuchi, F.; Takahashi, A.; Maeda, S.; Tsunoda, T.; Chen, P.; et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 2012, 44, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Wain, L.V.; Vaez, A.; Jansen, R.; Joehanes, R.; van der Most, P.J.; Erzurumluoglu, A.M.; O’Reilly, P.F.; Cabrera, C.P.; Warren, H.R.; Rose, L.M.; et al. Novel blood pressure locus and gene discovery using genome-wide association study and expression data sets from blood and the kidney. Hypertension 2017, 70, e4–e19. [Google Scholar] [CrossRef] [Green Version]
- Fehringer, G.; Kraft, P.; Pharoah, P.D.; Eeles, R.A.; Chatterjee, N.; Schumacher, F.R.; Schildkraut, J.M.; Lindström, S.; Brennan, P.; Bickeböller, H.; et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate, and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 2016, 76, 5103–5114. [Google Scholar] [CrossRef] [Green Version]
- Holder, A.M.; Akcakanat, A.; Adkins, F.; Evans, K.; Chen, H.; Wei, C.; Milton, D.R.; Li, Y.; Do, K.-A.; Janku, F.; et al. Epithelial to mesenchymal transition is associated with rapamycin resistance. Oncotarget 2015, 6, 19500–19513. [Google Scholar] [CrossRef] [Green Version]
- Valianou, M.; Filippidou, N.; Johnson, D.L.; Vogel, P.; Zhang, E.Y.; Liu, X.; Lu, Y.; Yu, J.J.; Bissler, J.J.; Astrinidis, A. Rapalog resistance is associated with mesenchymal-type changes in Tsc2-null cells. Sci. Rep. 2019, 9, 3015. [Google Scholar] [CrossRef]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I.; Benajiba, L.; Alexe, G.; Ramos, A.; Pikman, Y.; Conway, A.S.; Burgess, M.R.; Li, Q.; et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 2017, 23, 301–313. [Google Scholar] [CrossRef]
- Saito, Y.; Sawa, D.; Kinoshita, M.; Yamada, A.; Kamimura, S.; Suekane, A.; Ogoh, H.; Matsuo, H.; Adachi, S.; Taga, T.; et al. EVI1 triggers metabolic reprogramming associated with leukemogenesis and increases sensitivity to L-asparaginase. Haematologica 2019. [Google Scholar] [CrossRef]
- Sánchez-Gastaldo, A.; Kempf, E.; González Del Alba, A.; Duran, I. Systemic treatment of renal cell cancer: A comprehensive review. Cancer Treat. Rev. 2017, 60, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.T.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, L.; Stec, R.; Cierniak, S.; Synowiec, A.; Wcisło, G.; Jesiotr, M.; Koktysz, R.; Kozłowski, W.; Szczylik, C. Clinical usefulness of PI3K/Akt/mTOR genotyping in companion with other clinical variables in metastatic renal cell carcinoma patients treated with everolimus in the second and subsequent lines. Ann. Oncol. 2015, 26, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Sauerbrei, W.; Taube, S.E.; McShane, L.M.; Cavenagh, M.M.; Altman, D.G. Reporting recommendations for tumor marker prognostic studies (REMARK): an abridged explanation and elaboration. J. Natl. Cancer Inst. 2018, 110, 803–811. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, e313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomero, L.; Bodnar, L.; Mateo, F.; Herranz-Ors, C.; Espín, R.; García-Varelo, M.; Jesiotr, M.; Ruiz de Garibay, G.; Casanovas, O.; López, J.I.; et al. EVI1 as a Prognostic and Predictive Biomarker of Clear Cell Renal Cell Carcinoma. Cancers 2020, 12, 300. https://doi.org/10.3390/cancers12020300
Palomero L, Bodnar L, Mateo F, Herranz-Ors C, Espín R, García-Varelo M, Jesiotr M, Ruiz de Garibay G, Casanovas O, López JI, et al. EVI1 as a Prognostic and Predictive Biomarker of Clear Cell Renal Cell Carcinoma. Cancers. 2020; 12(2):300. https://doi.org/10.3390/cancers12020300
Chicago/Turabian StylePalomero, Luis, Lubomir Bodnar, Francesca Mateo, Carmen Herranz-Ors, Roderic Espín, Mar García-Varelo, Marzena Jesiotr, Gorka Ruiz de Garibay, Oriol Casanovas, José I. López, and et al. 2020. "EVI1 as a Prognostic and Predictive Biomarker of Clear Cell Renal Cell Carcinoma" Cancers 12, no. 2: 300. https://doi.org/10.3390/cancers12020300