Neural Networks Recapitulation by Cancer Cells Promotes Disease Progression: A Novel Role of p73 Isoforms in Cancer-Neuronal Crosstalk

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Phenotype-Driven Identification of Genes Essential for Normal Nervous System Development and Neurological Function
2.2. Nervous System-Related Genes Are Frequently Mutated in Cancer Patient Tumors and Enriched in Cancer Driver Genes
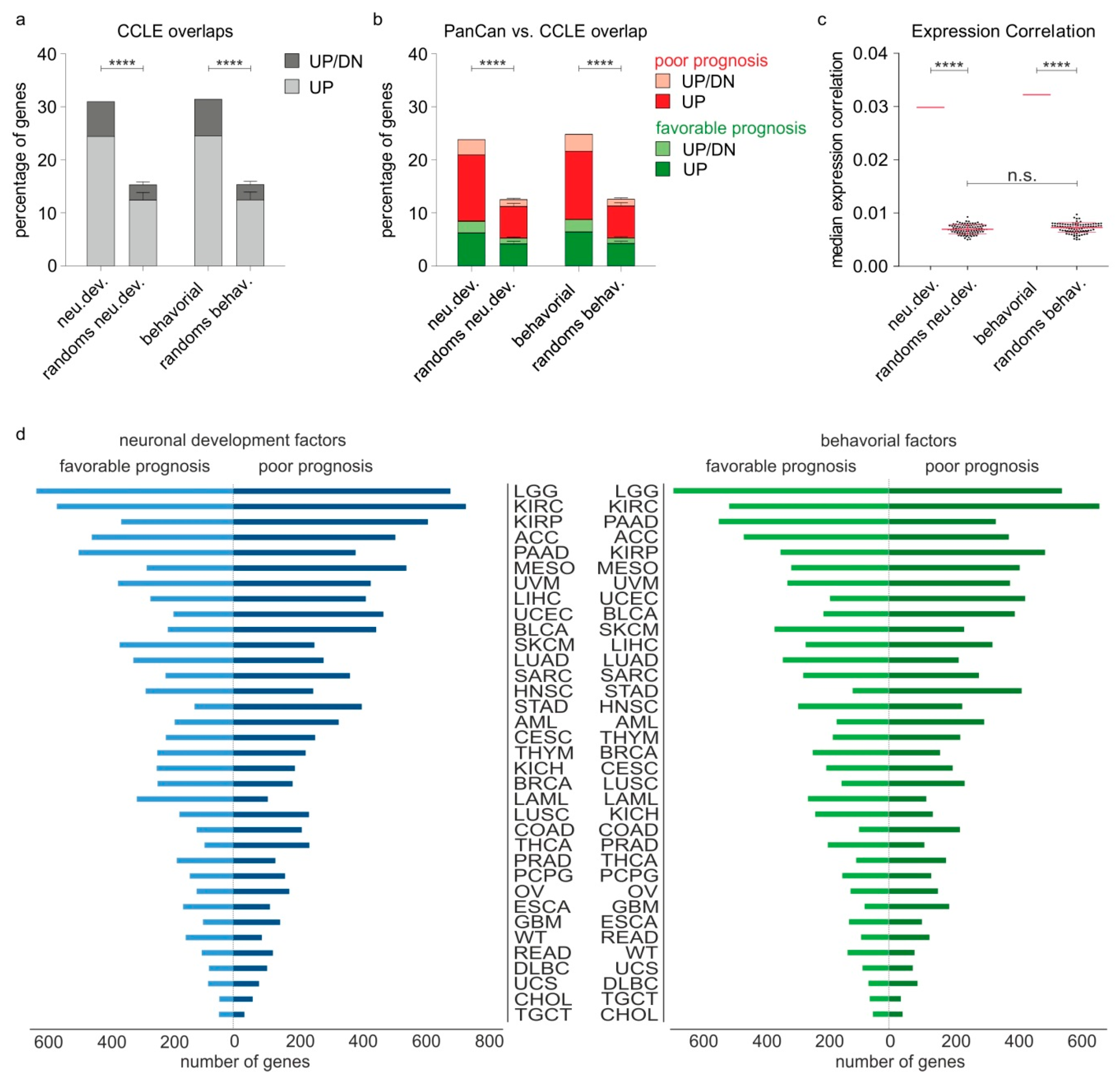
2.3. Nervous System-Related Genes Are Preferentially Upregulated in Aggressive Stages and Predict Patient Outcomes in a Cancer Type-Dependent Manner
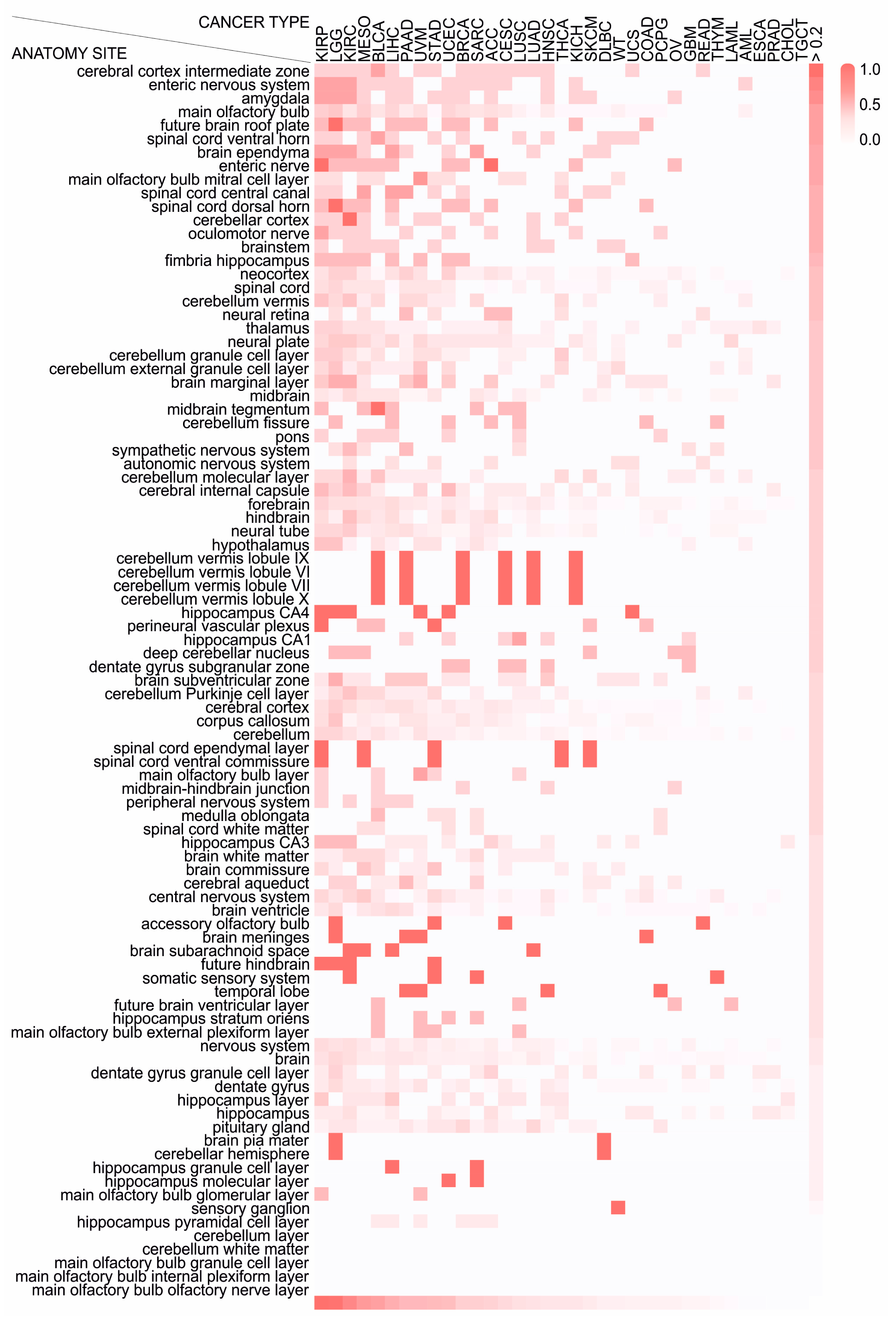
2.4. Different Cancer Types Co-Opt Genes Essential for the Development of Distinct Anatomical Parts of the Nervous System
2.5. Genes Involved in Several Stages of Neurogenesis and Neuronal Differentiation are Reactivated during Cancer Progression
2.6. Neurogenesis and Neoneurogenesis Mediators Are Abundantly Expressed during Melanoma Progression
2.7. Neurogenesis, Neoneurogenesis and Stemness Factors Are Concomitantly Regulated by TP73-Derived Isoforms
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Adenovector Generation and Transduction
4.3. RNA Isolation and q-PCR
4.4. Western Blot
4.5. Identification of Nervous System-Related Genes
4.6. Gene Expression Analysis in CCLE Database
4.7. Kaplan–Meier Survival Analysis of TCGA Patient Data
4.8. Neurodifferentiation Gene Expression Data Analysis
4.8.1. Transcriptomic Data Retrieval
4.8.2. Differential Gene Expression Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goody, D.; Gupta, S.K.; Engelmann, D.; Spitschak, A.; Marquardt, S.; Mikkat, S.; Meier, C.; Hauser, C.; Gundlach, J.P.; Egberts, J.H.; et al. Drug Repositioning Inferred from E2F1-Coregulator Interactions Studies for the Prevention and Treatment of Metastatic Cancers. Theranostics 2019, 9, 1490–1509. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; Patel, S.A.; Harewood, L.; Olan, I.; Vojtasova, E.; Syafruddin, S.E.; Zaini, M.N.; Richardson, E.K.; Burge, J.; Warren, A.Y.; et al. NF-κB-Dependent Lymphoid Enhancer Co-option Promotes Renal Carcinoma Metastasis. Cancer Discov. 2018, 8, 850–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 2013, 5, 186ra166. [Google Scholar] [CrossRef] [Green Version]
- Richter, C.; Marquardt, S.; Li, F.; Spitschak, A.; Murr, N.; Edelhäuser, B.A.H.; Iliakis, G.; Pützer, B.M.; Logotheti, S. Rewiring E2F1 with classical NHEJ via APLF suppression promotes bladder cancer invasiveness. J. Exp. Clin. Cancer Res. 2019, 38, 292. [Google Scholar] [CrossRef] [PubMed]
- Billaud, M.; Santoro, M. Is Co-option a prevailing mechanism during cancer progression? Cancer Res. 2011, 71, 6572–6575. [Google Scholar] [CrossRef] [Green Version]
- Delloye-Bourgeois, C.; Castellani, V. Hijacking of Embryonic Programs by Neural Crest-Derived Neuroblastoma: From Physiological Migration to Metastatic Dissemination. Front. Mol. Neurosci. 2019, 12, 52. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, V.; Bardelli, A.; Siena, S.; Abrignani, S. Exploring the links between cancer and placenta development. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Martik, M.L.; Bronner, M.E. Regulatory Logic Underlying Diversification of the Neural Crest. Trends Genet. 2017, 33, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.; Firlej, V.; Allory, Y.; Roméo, P.H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C. Role of the autonomic nervous system in tumorigenesis and metastasis. Mol. Cell. Oncol. 2015, 2, e975643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, M.; Colbert, P.L.; Vermeer, D.W.; Lucido, C.T.; Cain, J.T.; Vichaya, E.G.; Grossberg, A.J.; Muirhead, D.; Rickel, A.P.; Hong, Z.; et al. Cancer exosomes induce tumor innervation. Nat. Commun. 2018, 9, 4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta 2011, 1816, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Su, A.; Dry, S.M.; Binder, S.W.; Said, J.; Shintaku, P.; Sarantopoulos, G.P. Malignant melanoma with neural differentiation: An exceptional case report and brief review of the pertinent literature. Am. J. Dermatopathol. 2014, 36, e5–e9. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Fan, C.; Shangguan, W.; Liu, Y.; Li, Y.; Shang, Y.; Yin, D.; Zhang, S.; Huang, Q.; Li, X.; et al. Neurons generated from carcinoma stem cells support cancer progression. Signal Transduct. Target. Ther. 2017, 2, 16036. [Google Scholar] [CrossRef] [Green Version]
- Gregory, E.; Dugan, R.; David, G.; Song, Y.H. The biology and engineered modeling strategies of cancer-nerve crosstalk. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188406. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e318. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnucci, C.; Nizzardo, M.; Corti, S.; Zanni, G.; Bertini, E. In vitro neurogenesis: Development and functional implications of iPSC technology. Cell. Mol. Life Sci. 2014, 71, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, X.; Zack, D.J.; Zhu, H.; Qian, J. TiGER: A database for tissue-specific gene expression and regulation. BMC Bioinform. 2008, 9, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evergren, E.; Benfenati, F.; Shupliakov, O. The synapsin cycle: A view from the synaptic endocytic zone. J. Neurosci. Res. 2007, 85, 2648–2656. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments at a glance. J. Cell Sci. 2012, 125, 3257–3263. [Google Scholar] [CrossRef]
- Müller, T.; Braud, S.; Jüttner, R.; Voigt, B.C.; Paulick, K.; Sheean, M.E.; Klisch, C.; Gueneykaya, D.; Rathjen, F.G.; Geiger, J.R.; et al. Neuregulin 3 promotes excitatory synapse formation on hippocampal interneurons. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kakumoto, K.; Yoshida, T.; Kuwako, K.I.; Miyazaki, T.; Yamaguchi, J.; Konno, A.; Hata, J.; Uchiyama, Y.; Hirai, H.; et al. Elavl3 is essential for the maintenance of Purkinje neuron axons. Sci. Rep. 2018, 8, 2722. [Google Scholar] [CrossRef]
- Allen, J.K.; Armaiz-Pena, G.N.; Nagaraja, A.S.; Sadaoui, N.C.; Ortiz, T.; Dood, R.; Ozcan, M.; Herder, D.M.; Haemmerle, M.; Gharpure, K.M.; et al. Sustained Adrenergic Signaling Promotes Intratumoral Innervation through BDNF Induction. Cancer Res. 2018, 78, 3233–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 2665. [Google Scholar] [CrossRef] [PubMed]
- Steder, M.; Alla, V.; Meier, C.; Spitschak, A.; Pahnke, J.; Fürst, K.; Kowtharapu, B.S.; Engelmann, D.; Petigk, J.; Egberts, F.; et al. DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell 2013, 24, 512–527. [Google Scholar] [CrossRef] [Green Version]
- Logotheti, S.; Pavlopoulou, A.; Galtsidis, S.; Vojtesek, B.; Zoumpourlis, V. Functions, divergence and clinical value of TAp73 isoforms in cancer. Cancer Metastasis Rev. 2013, 32, 511–534. [Google Scholar] [CrossRef]
- Meier, C.; Hardtstock, P.; Joost, S.; Alla, V.; Pützer, B.M. p73 and IGF1R Regulate Emergence of Aggressive Cancer Stem-like Features via miR-885-5p Control. Cancer Res. 2016, 76, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Logotheti, S.; Marquardt, S.; Pützer, B.M. p73-Governed miRNA Networks: Translating Bioinformatics Approaches to Therapeutic Solutions for Cancer Metastasis. Methods Mol. Biol. 2019, 1912, 33–52. [Google Scholar] [CrossRef]
- Engelmann, D.; Pützer, B.M. Emerging from the shade of p53 mutants: N-terminally truncated variants of the p53 family in EMT signaling and cancer progression. Sci. Signal 2014, 7, re9. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, D.; Meier, C.; Alla, V.; Pützer, B.M. A balancing act: Orchestrating amino-truncated and full-length p73 variants as decisive factors in cancer progression. Oncogene 2015, 34, 4287–4299. [Google Scholar] [CrossRef] [Green Version]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Killick, R.; Niklison-Chirou, M.; Tomasini, R.; Bano, D.; Rufini, A.; Grespi, F.; Velletri, T.; Tucci, P.; Sayan, B.S.; Conforti, F.; et al. p73: A multifunctional protein in neurobiology. Mol. Neurobiol. 2011, 43, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemajerova, A.; Amelio, I.; Gebel, J.; Dötsch, V.; Melino, G.; Moll, U.M. Non-oncogenic roles of TAp73: From multiciliogenesis to metabolism. Cell Death Differ. 2018, 25, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Niklison-Chirou, M.V.; Agostini, M.; Amelio, I.; Melino, G. Regulation of Adult Neurogenesis in Mammalian Brain. Int. J. Mol. Sci. 2020, 21, 4869. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K. p73: A Positive or Negative Regulator of Angiogenesis, or Both? Mol. Cell. Biol. 2015, 36, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Sharif, T.; Dai, C.; Martell, E.; Ghassemi-Rad, M.S.; Hanes, M.R.; Murphy, P.J.; Kennedy, B.E.; Venugopal, C.; Subapanditha, M.; Giacomantonio, C.A.; et al. TAp73 Modifies Metabolism and Positively Regulates Growth of Cancer Stem-Like Cells in a Redox-Sensitive Manner. Clin. Cancer Res. 2019, 25, 2001–2017. [Google Scholar] [CrossRef]
- Galtsidis, S.; Logotheti, S.; Pavlopoulou, A.; Zampetidis, C.P.; Papachristopoulou, G.; Scorilas, A.; Vojtesek, B.; Gorgoulis, V.; Zoumpourlis, V. Unravelling a p73-regulated network: The role of a novel p73-dependent target, MIR3158, in cancer cell migration and invasiveness. Cancer Lett. 2017, 388, 96–106. [Google Scholar] [CrossRef]
- Hinske, L.C.; França, G.S.; Torres, H.A.; Ohara, D.T.; Lopes-Ramos, C.M.; Heyn, J.; Reis, L.F.; Ohno-Machado, L.; Kreth, S.; Galante, P.A. miRIAD-integrating microRNA inter- and intragenic data. Database (Oxford) 2014, 2014. [Google Scholar] [CrossRef]
- Fürst, K.; Steder, M.; Logotheti, S.; Angerilli, A.; Spitschak, A.; Marquardt, S.; Schumacher, T.; Engelmann, D.; Herchenröder, O.; Rupp, R.A.W.; et al. DNp73-induced degradation of tyrosinase links depigmentation with EMT-driven melanoma progression. Cancer Lett. 2019, 442, 299–309. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; García-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef]
- Kozlov, E.M.; Grechko, A.V.; Chegodaev, Y.S.; Wu, W.K.; Orekhov, A.N. Contribution of Neurotrophins to the Immune System Regulation and Possible Connection to Alcohol Addiction. Biology 2020, 9, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiewe, T.; Theseling, C.C.; Pützer, B.M. Transactivation-deficient Delta TA-p73 inhibits p53 by direct competition for DNA binding: Implications for tumorigenesis. J. Biol. Chem. 2002, 277, 14177–14185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, K.A.; Yates, B.; Seal, R.L.; Wright, M.W.; Bruford, E.A. Genenames.org: The HGNC resources in 2015. Nucleic Acids Res. 2015, 43, D1079–D1085. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Athar, A.; Fullgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Parkinson, H.; Sarkans, U.; Kolesnikov, N.; Abeygunawardena, N.; Burdett, T.; Dylag, M.; Emam, I.; Farne, A.; Hastings, E.; Holloway, E.; et al. ArrayExpress update—An archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011, 39, D1002–D1004. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Liu, J.; Sells Galvin, R.J.; Dage, J.L.; Egeland, J.A.; Smith, R.C.; Merchant, K.M.; Paul, S.M. Transcriptomic Analysis of Induced Pluripotent Stem Cells Derived from Patients with Bipolar Disorder from an Old Order Amish Pedigree. PLoS ONE 2015, 10, e0142693. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.; Sommer, A.; Plotz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Campanac, E.; Cheung, H.H.; Ziats, M.N.; Canterel-Thouennon, L.; Raygada, M.; Baxendale, V.; Pang, A.L.; Yang, L.; Swedo, S.; et al. Idiopathic Autism: Cellular and Molecular Phenotypes in Pluripotent Stem Cell-Derived Neurons. Mol. Neurobiol. 2017, 54, 4507–4523. [Google Scholar] [CrossRef]
- Calzari, L.; Barcella, M.; Alari, V.; Braga, D.; Munoz-Viana, R.; Barlassina, C.; Finelli, P.; Gervasini, C.; Barco, A.; Russo, S.; et al. Transcriptome Analysis of iPSC-Derived Neurons from Rubinstein-Taybi Patients Reveals Deficits in Neuronal Differentiation. Mol. Neurobiol. 2020, 57, 3685–3701. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Logotheti, S.; Marquardt, S.; Richter, C.; Sophie Hain, R.; Murr, N.; Takan, I.; Pavlopoulou, A.; Pützer, B.M. Neural Networks Recapitulation by Cancer Cells Promotes Disease Progression: A Novel Role of p73 Isoforms in Cancer-Neuronal Crosstalk. Cancers 2020, 12, 3789. https://doi.org/10.3390/cancers12123789
Logotheti S, Marquardt S, Richter C, Sophie Hain R, Murr N, Takan I, Pavlopoulou A, Pützer BM. Neural Networks Recapitulation by Cancer Cells Promotes Disease Progression: A Novel Role of p73 Isoforms in Cancer-Neuronal Crosstalk. Cancers. 2020; 12(12):3789. https://doi.org/10.3390/cancers12123789
Chicago/Turabian StyleLogotheti, Stella, Stephan Marquardt, Christin Richter, Renée Sophie Hain, Nico Murr, Işıl Takan, Athanasia Pavlopoulou, and Brigitte M. Pützer. 2020. "Neural Networks Recapitulation by Cancer Cells Promotes Disease Progression: A Novel Role of p73 Isoforms in Cancer-Neuronal Crosstalk" Cancers 12, no. 12: 3789. https://doi.org/10.3390/cancers12123789