Cancer Immunotherapy and Application of Nanoparticles in Cancers Immunotherapy as the Delivery of Immunotherapeutic Agents and as the Immunomodulators



Abstract
:Simple Summary
Abstract
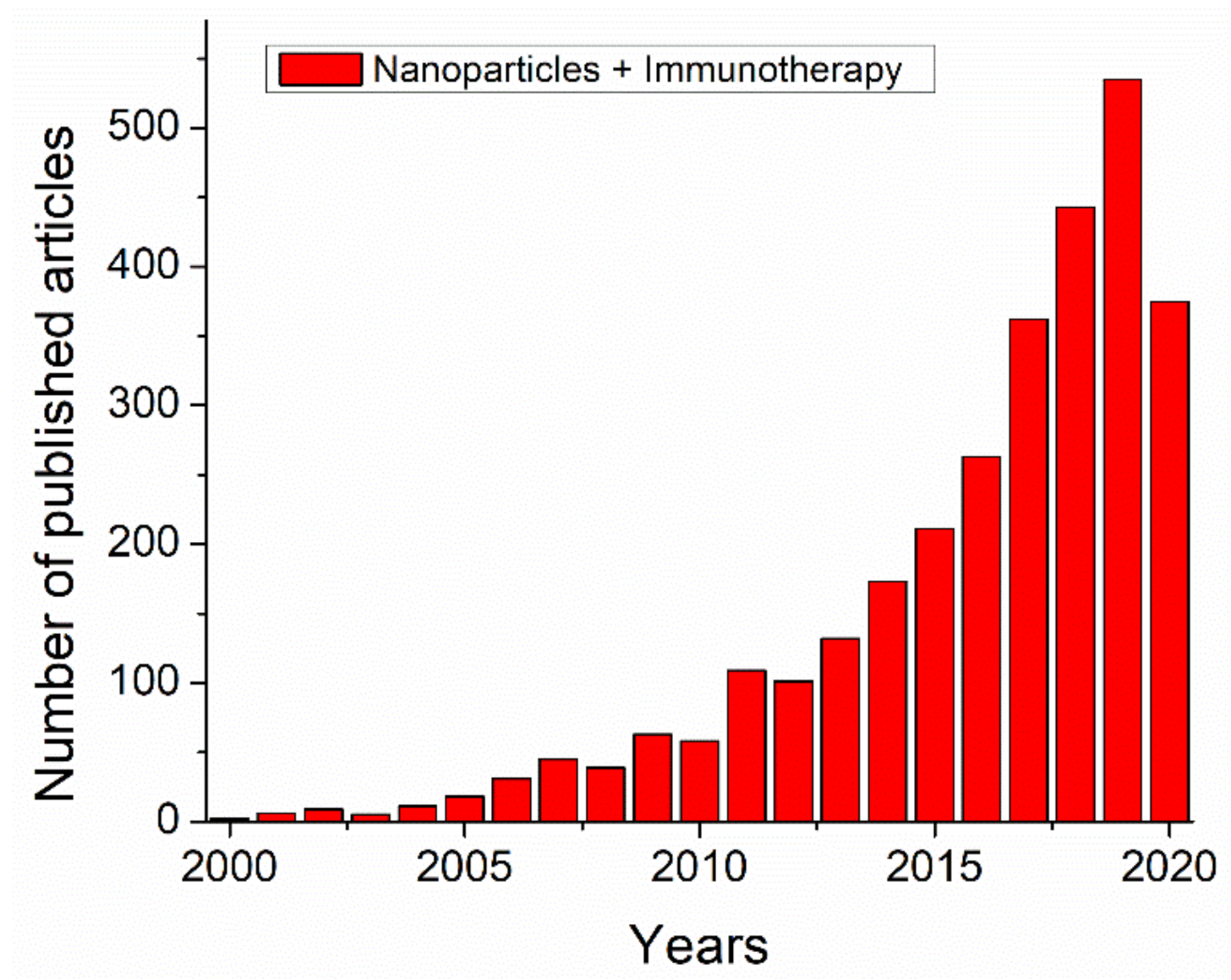
1. Introduction
2. Nanoparticles and Nanoparticles-Based Drug Delivery Systems
2.1. The Application of Nanoparticles in Cancer Immunotherapy
2.1.1. Nanoparticles as the Carrier of Immunotherapeutic Agents
2.1.2. Antigens and Adjuvants Delivery to Antigen Presenting Cells (APCs)
2.1.3. Antigens and Adjuvants Delivery to Tumor Microenvironment (TME)
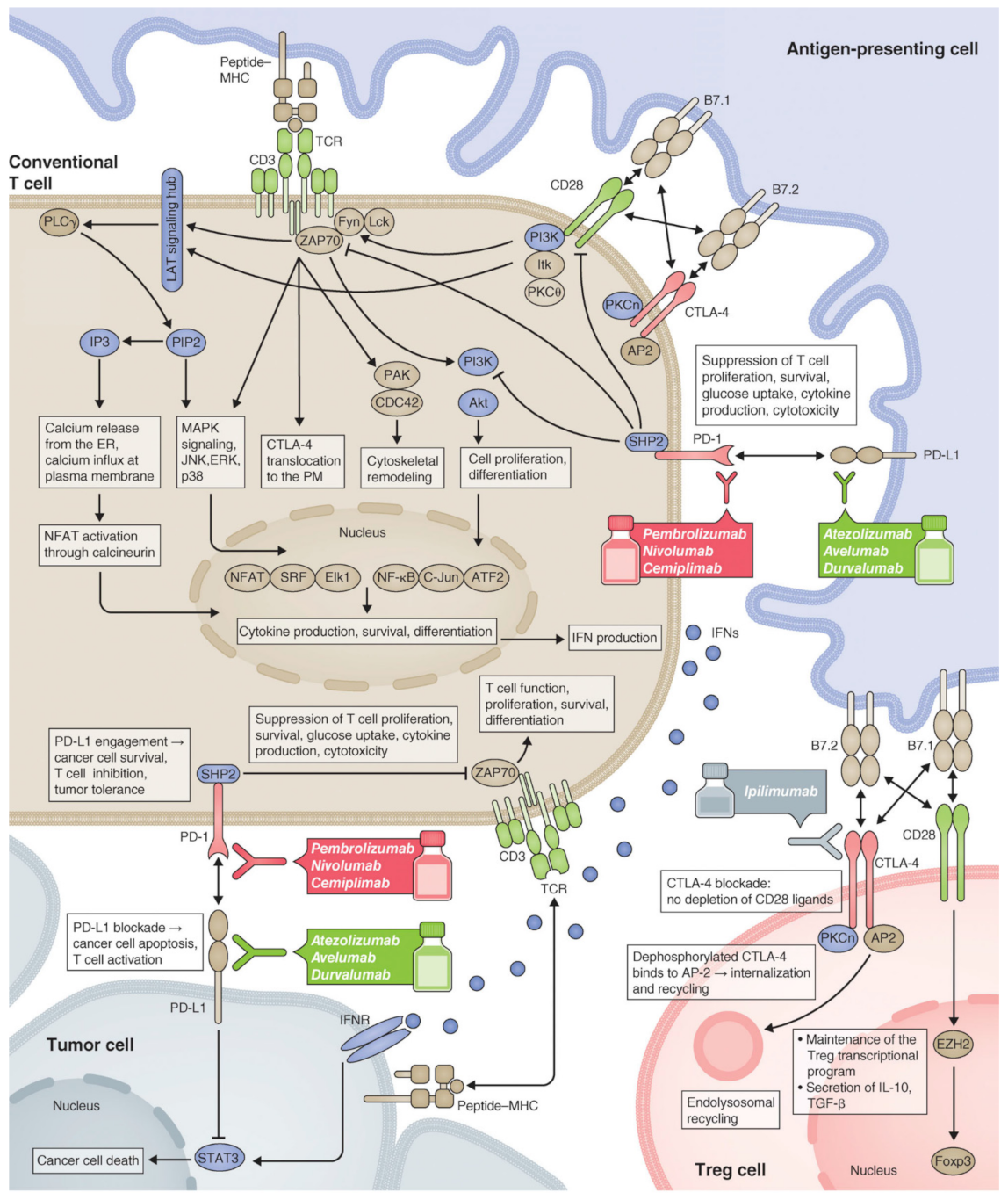
2.1.4. Immune Checkpoint Inhibitors (ICIs) Delivery
2.1.5. Nanoparticles as the Direct Immunomodulators
3. Clinical Translation of Nano-Immunotherapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McCormack, V.A.; Boffetta, P. Today’s lifestyles, tomorrow’s cancers: Trends in lifestyle risk factors for cancer in low- and middle-income countries. Ann. Oncol. 2011, 22, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Blackadar, C.B. Historical review of the causes of cancer. World J. Clin. Oncol. 2016, 7, 54–86. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [Green Version]
- Suo, X.; Zhang, J.; Zhang, Y.; Liang, X.J.; Zhang, J.; Liu, D. A nano-based thermotherapy for cancer stem cell-targeted therapy. J. Mater. Chem. B 2020, 8, 3985–4001. [Google Scholar] [CrossRef]
- Yin, P.T.; Shah, S.; Pasquale, N.J.; Garbuzenko, O.B.; Minko, T.; Lee, K.B. Stem cell-based gene therapy activated using magnetic hyperthermia to enhance the treatment of cancer. Biomaterials 2016, 81, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Spring, B.Q.; Rizvi, I.; Xu, N.; Hasan, T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem. Photobiol. Sci. 2015, 14, 1476–1491. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Bonvin, D.; Berndsen, R.H.; Scherrer, E.; Wong, T.J.; Dyson, P.J.; Griffioen, A.W.; Nowak-Sliwinska, P. Angiostatic treatment prior to chemo- or photodynamic therapy improves anti-tumor efficacy. Sci. Rep. 2015, 5, 8990. [Google Scholar] [CrossRef] [Green Version]
- Samant, R.S.; Shevde, L.A. Recent advances in anti-angiogenic therapy of cancer. Oncotarget 2011, 2, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.L. Biologic therapies: What and when? J. Clin. Pathol. 2007, 60, 8–17. [Google Scholar] [CrossRef]
- Cairns, R.; Papandreou, I.; Denko, N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumor microenvironment. Mol. Cancer Res. 2006, 4, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debele, T.A.; Mekuria, S.L.; Tsai, H.C. Polysaccharide based nanogels in the drug delivery system: Application as the carrier of pharmaceutical agents. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 68, 964–981. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. SMANCS and polymer-conjugated macromolecular drugs: Advantages in cancer chemotherapy. Adv. Drug Deliv. Rev. 2001, 46, 169–185. [Google Scholar] [CrossRef]
- Leu, A.J.; Berk, D.A.; Lymboussaki, A.; Alitalo, K.; Jain, R.K. Absence of functional lymphatics within a murine sarcoma: A molecular and functional evaluation. Cancer Res. 2000, 60, 4324–4327. [Google Scholar]
- Maeda, H. Tumor-Selective Delivery of Macromolecular Drugs via the EPR Effect: Background and Future Prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Debele, T.A.; Peng, S.; Tsai, H.-C. Drug Carrier for Photodynamic Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 22094–22136. [Google Scholar] [CrossRef]
- Chidambaram, M.; Manavalan, R.; Kathiresan, K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J. Pharm. Pharm. Sci. 2011, 14, 67–77. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Rosfa, S.; Wlodarski, A.; Kuharev, J.; Rekik, A.; Knauer, S.K.; Bantz, C.; Nawroth, T.; Bier, C.; et al. Nanoparticle size is a critical physicochemical determinant of the human blood plasma corona: A comprehensive quantitative proteomic analysis. ACS Nano 2011, 5, 7155–7167. [Google Scholar] [CrossRef]
- Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics 2019, 9, 126–151. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G.; Callipo, L.; De Sanctis, S.C.; Cavaliere, C.; Pozzi, D.; Laganà, A. Surface adsorption of protein corona controls the cell internalization mechanism of DC-Chol–DOPE/DNA lipoplexes in serum. Biochim. Biophys. Acta (BBA) Biomembranes 2010, 1798, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetti, C.; Motamedchaboki, K.; Magrini, A.; Palmieri, G.; Mattei, M.; Bernardini, S.; Rosato, N.; Bottini, N.; Bottini, M. Surface polyethylene glycol conformation influences the protein corona of polyethylene glycol-modified single-walled carbon nanotubes: Potential implications on biological performance. ACS Nano 2013, 7, 1974–1989. [Google Scholar] [CrossRef]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernández, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef]
- Moore, A.; Marecos, E.; Bogdanov, A., Jr.; Weissleder, R. Tumoral distribution of long-circulating dextran-coated iron oxide nanoparticles in a rodent model. Radiology 2000, 214, 568–574. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomedicine 2013, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-X.J. Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant. Imaging Med. Surg. 2011, 1, 35–40. [Google Scholar] [CrossRef]
- Kendall, M.; Lynch, I. Long-term monitoring for nanomedicine implants and drugs. Nat. Nanotechnol. 2016, 11, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia Pac. J. Oncol. Nurs. 2017, 4, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.Y.; Huang, L. Cancer immunotherapy and nanomedicine. Pharm. Res. 2011, 28, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gambhir, V.; Alatery, A.; Basta, S. Delivery of Exogenous Antigens to Induce Cytotoxic CD8+ T Lymphocyte Responses. J. Biomed. Biotechnol. 2010, 2010, 218752. [Google Scholar] [CrossRef]
- Shresta, S.; Pham, C.T.; Thomas, D.A.; Graubert, T.A.; Ley, T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998, 10, 581–587. [Google Scholar] [CrossRef]
- Basta, S.; Alatery, A. The Cross-priming Pathway: A Portrait of an Intricate Immune System. Scand. J. Immunol. 2007, 65, 311–319. [Google Scholar] [CrossRef]
- Bhat, P.; Leggatt, G.; Waterhouse, N.; Frazer, I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017, 8, e2836. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Frankel, S.R.; Baeuerle, P.A. Targeting T cells to tumor cells using bispecific antibodies. Curr. Opin. Chem. Biol. 2013, 17, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Fan, D.; Xiong, D. The development of bispecific antibodies and their applications in tumor immune escape. Exp. Hematol. Oncol. 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Ring, S.; Umansky, V.; Mahnke, K.; Enk, A.H. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J. Investig. Dermatol. 2012, 132, 1239–1246. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Onizuka, S.; Tawara, I.; Shimizu, J.; Sakaguchi, S.; Fujita, T.; Nakayama, E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999, 59, 3128–3133. [Google Scholar]
- Yang, L.; Edwards, C.M.; Mundy, G.R. Gr-1+CD11b+ myeloid-derived suppressor cells: Formidable partners in tumor metastasis. J. Bone Miner. Res. 2010, 25, 1701–1706. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Viñals, F.; Capellá, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Dimberu, P.M.; Leonhardt, R.M. Cancer immunotherapy takes a multi-faceted approach to kick the immune system into gear. Yale J. Biol. Med. 2011, 84, 371–380. [Google Scholar] [PubMed]
- Fang, L.; Lonsdorf, A.S.; Hwang, S.T. Immunotherapy for advanced melanoma. J. Investig. Dermatol. 2008, 128, 2596–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Atkins, M.B.; Subedi, P.; Wu, J.; Chambers, J.; Joseph Mattingly, T.; Campbell, J.D.; Allen, J.; Ferris, A.E.; Schilsky, R.L.; et al. The promise of Immuno-oncology: Implications for defining the value of cancer treatment. J. Immunother. Cancer 2019, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W.; et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1747. [Google Scholar] [CrossRef]
- Mohan, T.; Verma, P.; Rao, D.N. Novel adjuvants & delivery vehicles for vaccines development: A road ahead. Indian J. Med. Res. 2013, 138, 779–795. [Google Scholar] [PubMed]
- Trovato, M.; De Berardinis, P. Novel antigen delivery systems. World J. Virol. 2015, 4, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Grippin, A.J.; Sayour, E.J.; Mitchell, D.A. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology 2017, 6, e1290036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticles for nasal vaccination. Adv. Drug Deliv. Rev. 2009, 61, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, J.A.; Thomas, S.N.; Swartz, M.A. Materials engineering for immunomodulation. Nature 2009, 462, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Lu, W.-Y. Recent advances in lymphatic targeted drug delivery system for tumor metastasis. Cancer Biol. Med. 2014, 11, 247–254. [Google Scholar] [CrossRef]
- McLennan, D.N.; Porter, C.J.H.; Charman, S.A. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov. Today Technol. 2005, 2, 89–96. [Google Scholar] [CrossRef]
- Moon, J.J.; Huang, B.; Irvine, D.J. Engineering nano- and microparticles to tune immunity. Adv. Mater. 2012, 24, 3724–3746. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.J.; Hanson, M.C.; Rakhra, K.; Tokatlian, T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015, 115, 11109–11146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtis, I.C.; Hirosue, S.; de Titta, A.; Kontos, S.; Stegmann, T.; Hubbell, J.A.; Swartz, M.A. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS ONE 2013, 8, e61646. [Google Scholar] [CrossRef]
- Mueller, S.N.; Tian, S.; DeSimone, J.M. Rapid and Persistent Delivery of Antigen by Lymph Node Targeting PRINT Nanoparticle Vaccine Carrier To Promote Humoral Immunity. Mol. Pharm. 2015, 12, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Tran, K.K.; Shen, H. Effect of the Poly(ethylene glycol) (PEG) Density on the Access and Uptake of Particles by Antigen-Presenting Cells (APCs) after Subcutaneous Administration. Mol. Pharm. 2012, 9, 3442–3451. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef] [PubMed]
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgdorf, S.; Kautz, A.; Böhnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Hirosue, S.; Hubbell, J.A. Engineering approaches to immunotherapy. Sci. Transl. Med. 2012, 4, 148rv9. [Google Scholar] [CrossRef]
- Shen, H.; Ackerman, A.L.; Cody, V.; Giodini, A.; Hinson, E.R.; Cresswell, P.; Edelson, R.L.; Saltzman, W.M.; Hanlon, D.J. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117, 78–88. [Google Scholar] [CrossRef] [PubMed]
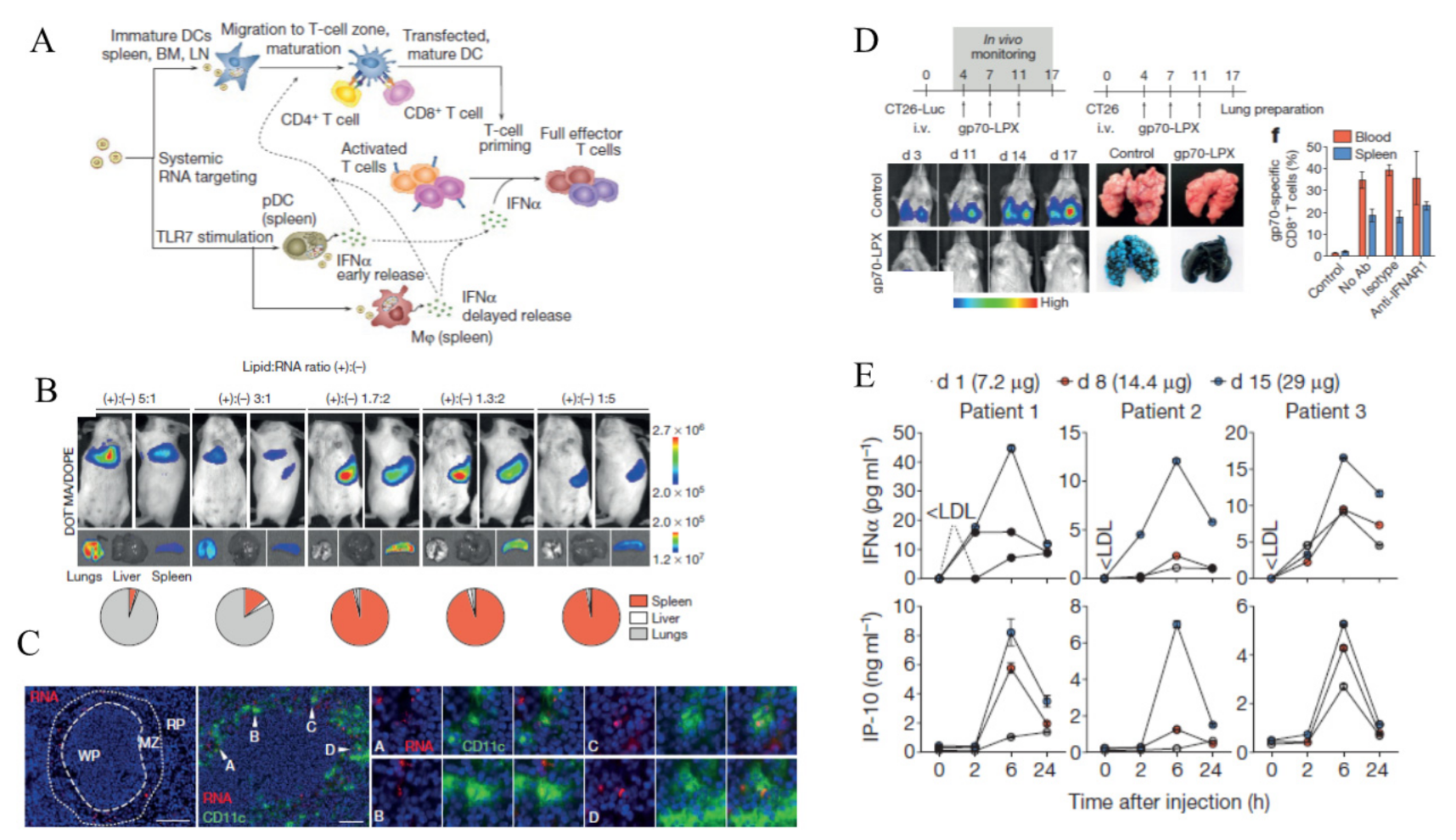
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Wille-Reece, U.; Flynn, B.J.; Loré, K.; Koup, R.A.; Kedl, R.M.; Mattapallil, J.J.; Weiss, W.R.; Roederer, M.; Seder, R.A. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA 2005, 102, 15190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, A.; Fine, R.L.; Demento, S.; Bockenstedt, L.K.; Fahmy, T.M. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials 2011, 32, 3094–3105. [Google Scholar] [CrossRef] [Green Version]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Bozzacco, L.; Trumpfheller, C.; Siegal, F.P.; Mehandru, S.; Markowitz, M.; Carrington, M.; Nussenzweig, M.C.; Piperno, A.G.; Steinman, R.M. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc. Natl. Acad. Sci. USA 2007, 104, 1289. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Rosalia, R.A.; Kleinovink, J.W.; Rueda, F.; Löwik, C.W.G.M.; Ossendorp, F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: A comparative study. J. Control. Release 2014, 192, 209–218. [Google Scholar] [CrossRef]
- Schlosser, E.; Mueller, M.; Fischer, S.; Basta, S.; Busch, D.H.; Gander, B.; Groettrup, M. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine 2008, 26, 1626–1637. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Kandalaft, L.E.; Coukos, G. Adjuvants for enhancing the immunogenicity of whole tumor cell vaccines. Int. Rev. Immunol. 2011, 30, 150–182. [Google Scholar] [CrossRef]
- Soiffer, R.; Hodi, F.S.; Haluska, F.; Jung, K.; Gillessen, S.; Singer, S.; Tanabe, K.; Duda, R.; Mentzer, S.; Jaklitsch, M.; et al. Vaccination With Irradiated, Autologous Melanoma Cells Engineered to Secrete Granulocyte-Macrophage Colony-Stimulating Factor by Adenoviral-Mediated Gene Transfer Augments Antitumor Immunity in Patients With Metastatic Melanoma. J. Clin. Oncol. 2003, 21, 3343–3350. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, F.; Ni, Q.; Niu, G.; Chen, X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano 2017, 11, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]
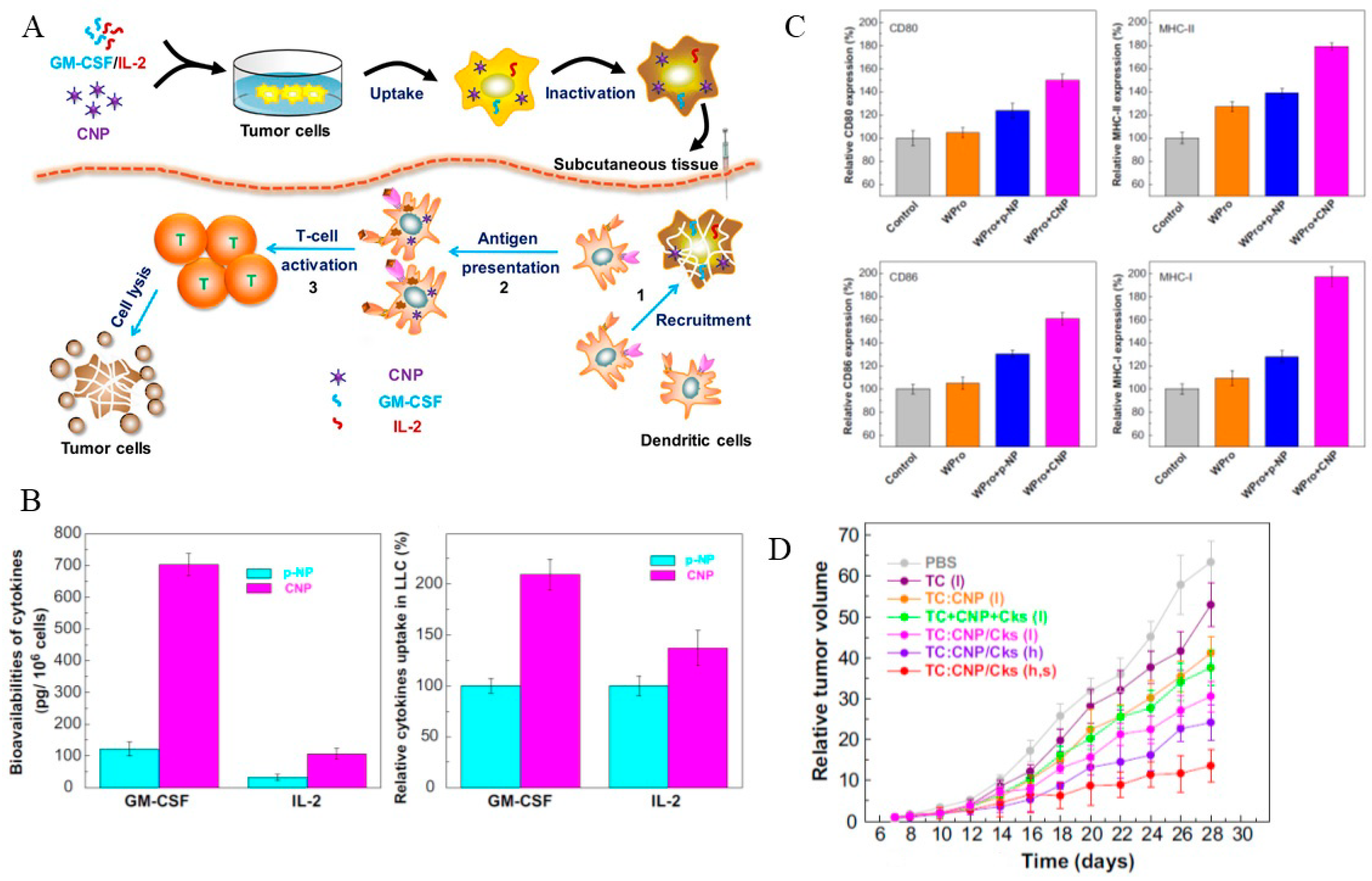
- Liu, S.-Y.; Wei, W.; Yue, H.; Ni, D.-Z.; Yue, Z.-G.; Wang, S.; Fu, Q.; Wang, Y.-Q.; Ma, G.-H.; Su, Z.-G. Nanoparticles-based multi-adjuvant whole cell tumor vaccine for cancer immunotherapy. Biomaterials 2013, 34, 8291–8300. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef]
- Mandapathil, M.; Szczepanski, M.J.; Szajnik, M.; Ren, J.; Jackson, E.K.; Johnson, J.T.; Gorelik, E.; Lang, S.; Whiteside, T.L. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J. Biol. Chem. 2010, 285, 27571–27580. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Godin-Ethier, J.; Hanafi, L.A.; Piccirillo, C.A.; Lapointe, R. Indoleamine 2,3-dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, H.; Cibrián, D.; Sánchez-Madrid, F. Immunoregulatory molecules are master regulators of inflammation during the immune response. FEBS Lett. 2012, 586, 2897–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Yang, B.; Gao, T.; Liu, T.; Li, J. Polymer nanoparticle-assisted chemotherapy of pancreatic cancer. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetti, C.; Rapini, N.; Magrini, A.; Cirelli, E.; Bellucci, S.; Mattei, M.; Rosato, N.; Bottini, N.; Bottini, M. In Vivo Targeting of Intratumor Regulatory T Cells Using PEG-Modified Single-Walled Carbon Nanotubes. Bioconjugate Chem. 2013, 24, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Niu, M.; O’Mary, H.; Cui, Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol. Pharm. 2013, 10, 3525–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
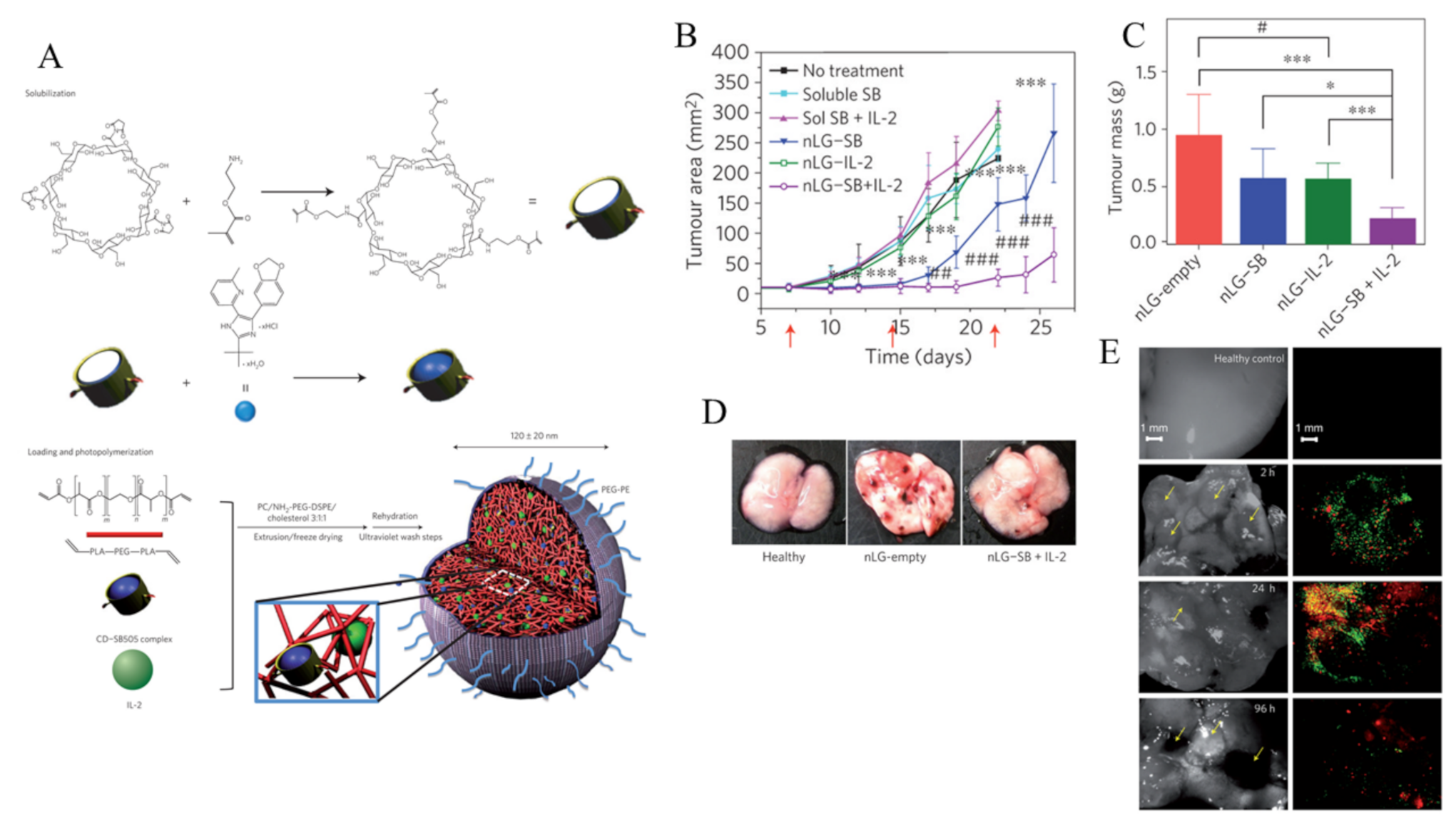
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069. [Google Scholar] [CrossRef] [Green Version]
- Riva, A.; Chokshi, S. Immune checkpoint receptors: Homeostatic regulators of immunity. Hepatol. Int. 2018, 12, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, D.M. A Nobel Prize-worthy pursuit: Cancer immunology and harnessing immunity to tumour neoantigens. Immunology 2018, 155, 283–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.; Arkenau, H.-T.; Infante, J.R. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs 2014, 74, 1993–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E. Immune checkpoint inhibitors. J. Cell Biol. 2019, 218, 740–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
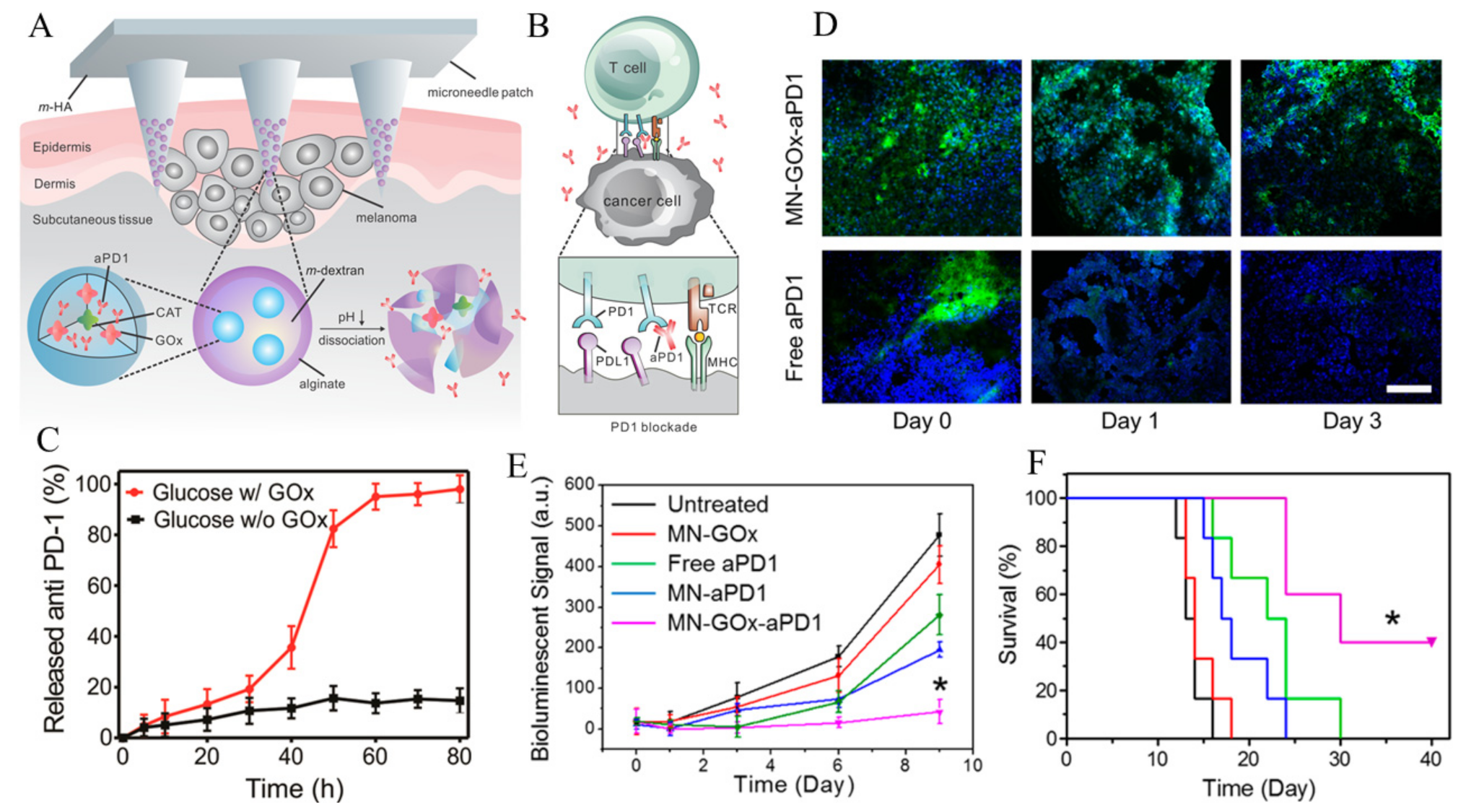
- Wang, C.; Ye, Y.; Hochu, G.M.; Sadeghifar, H.; Gu, Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16, 2334–2340. [Google Scholar] [CrossRef]
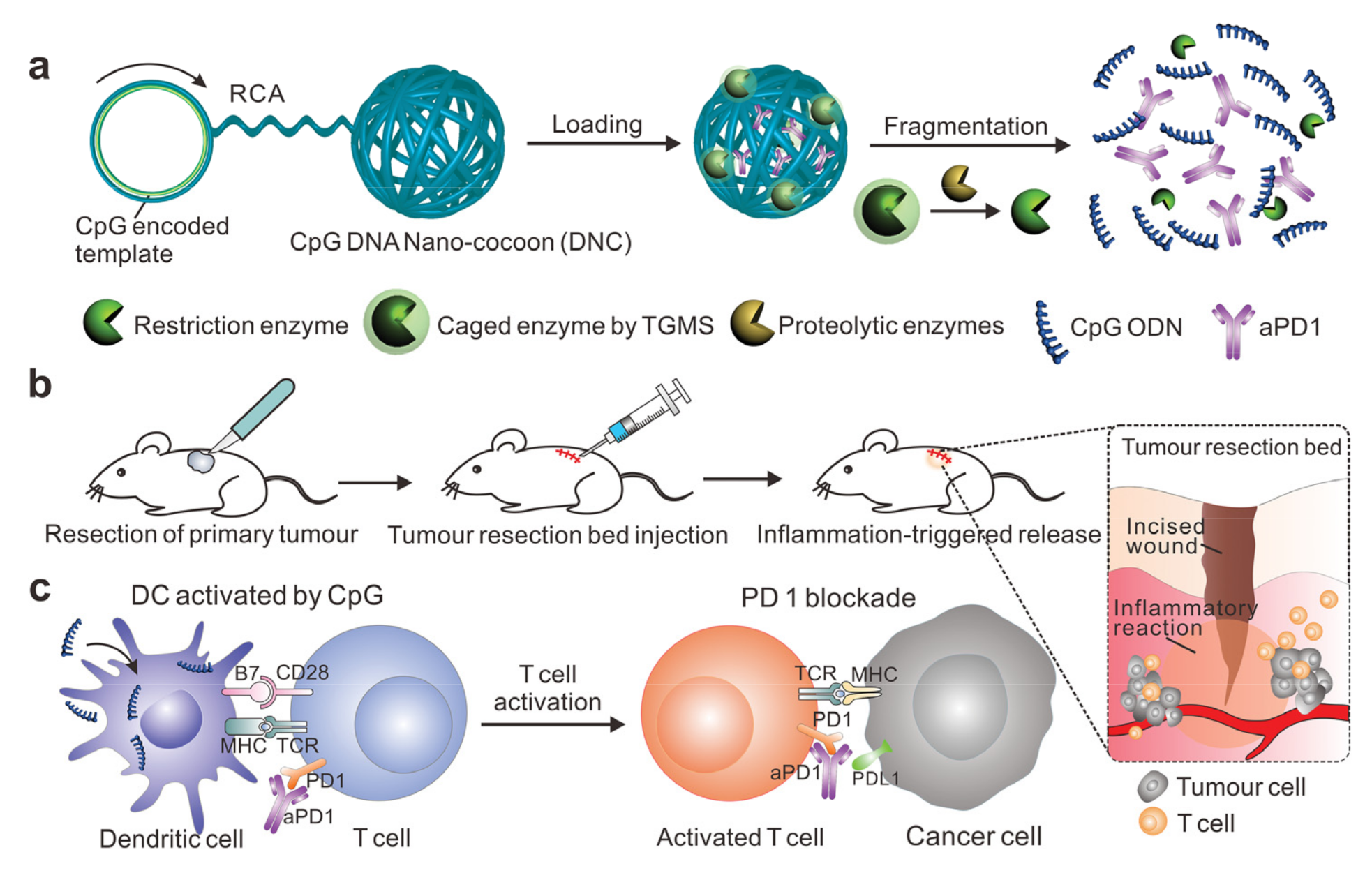
- Wang, C.; Sun, W.; Wright, G.; Wang, A.Z.; Gu, Z. Inflammation-Triggered Cancer Immunotherapy by Programmed Delivery of CpG and Anti-PD1 Antibody. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Héninger, E.; Krueger, T.E.G.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Hu, Q.; Wen, D.; Chen, Q.; Chen, G.; Lu, Y.; Wang, J.; Cheng, H.; Lu, W.; Gu, Z. A Dual-Bioresponsive Drug-Delivery Depot for Combination of Epigenetic Modulation and Immune Checkpoint Blockade. Adv. Mater. 2019, 31, e1806957. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Roche, K.C.; Tian, S.; Eblan, M.J.; McKinnon, K.P.; Caster, J.M.; Chai, S.; Herring, L.E.; Zhang, L.; Zhang, T.; et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 877–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palena, C.; Abrams, S.I.; Schlom, J.; Hodge, J.W. Cancer vaccines: Preclinical studies and novel strategies. Adv. Cancer Res. 2006, 95, 115–145. [Google Scholar] [CrossRef] [PubMed]
- Ogi, C.; Aruga, A. Immunological monitoring of anticancer vaccines in clinical trials. Oncoimmunology 2013, 2, e26012. [Google Scholar] [CrossRef] [Green Version]
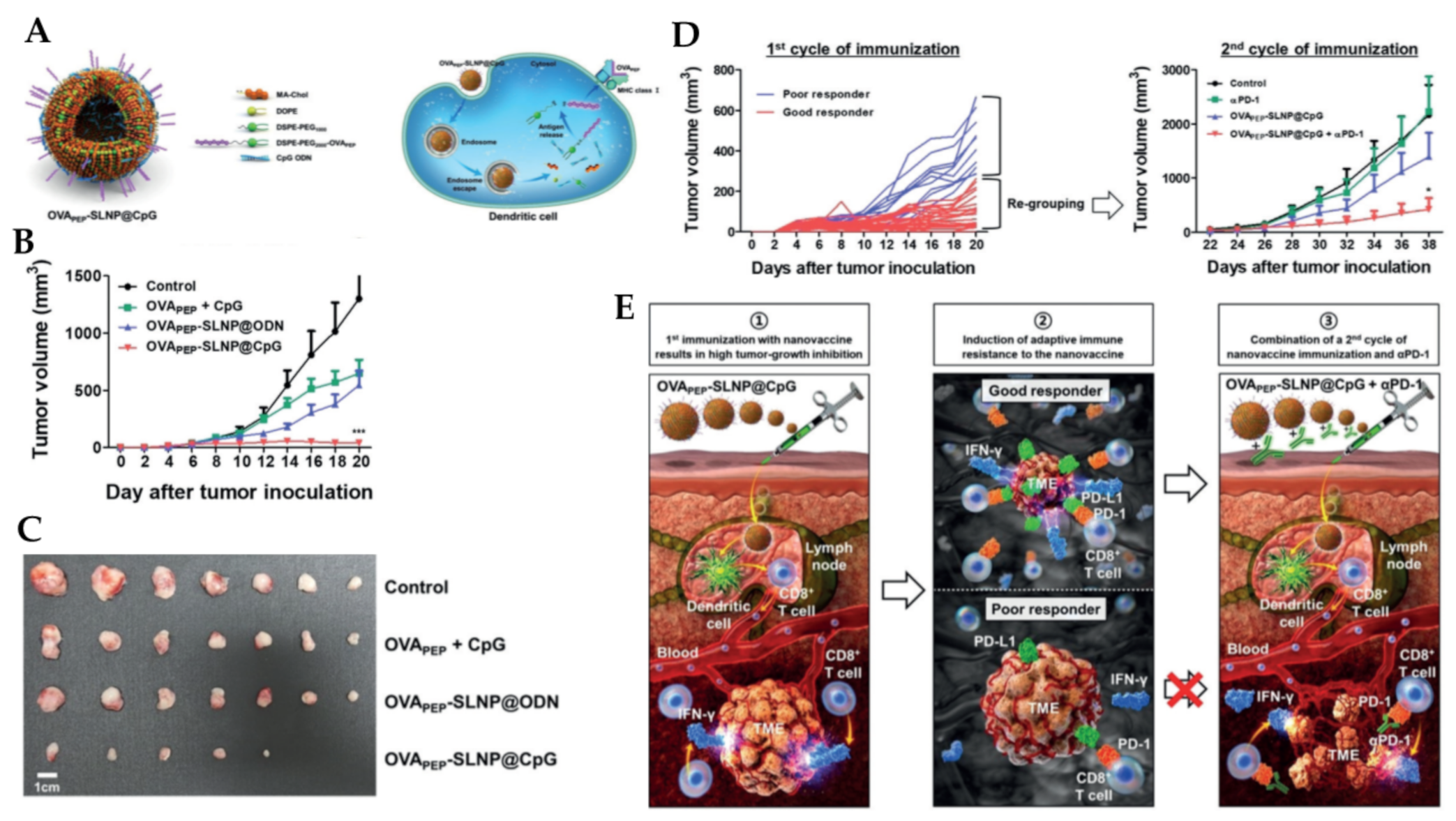
- Kim, Y.; Kang, S.; Shin, H.; Kim, T.; Yu, B.; Kim, J.; Yoo, D.; Jon, S. Sequential and Timely Combination of a Cancer Nanovaccine with Immune Checkpoint Blockade Effectively Inhibits Tumor Growth and Relapse. Angew. Chem. Int. Ed. 2020, 59, 14628–14638. [Google Scholar] [CrossRef]
- Fontana, F.; Fusciello, M.; Groeneveldt, C.; Capasso, C.; Chiaro, J.; Feola, S.; Liu, Z.; Mäkilä, E.M.; Salonen, J.J.; Hirvonen, J.T.; et al. Biohybrid Vaccines for Improved Treatment of Aggressive Melanoma with Checkpoint Inhibitor. ACS Nano 2019, 13, 6477–6490. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Holay, M.; Park, J.H.; Fang, R.H.; Zhang, J.; Zhang, L. Nanoparticle Delivery of Immunostimulatory Agents for Cancer Immunotherapy. Theranostics 2019, 9, 7826–7848. [Google Scholar] [CrossRef]
- Sushnitha, M.; Evangelopoulos, M.; Tasciotti, E.; Taraballi, F. Cell Membrane-Based Biomimetic Nanoparticles and the Immune System: Immunomodulatory Interactions to Therapeutic Applications. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Getts, D.R.; Shea, L.D.; Miller, S.D.; King, N.J. Harnessing nanoparticles for immune modulation. Trends Iimmunol. 2015, 36, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Q.; Li, L.; Mu, Q.; Zhang, Q. Immunomodulation of nanoparticles in nanomedicine applications. BioMed Res. Int. 2014, 2014, 426028. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Q.; Tang, R.-Z. Biological responses to nanomaterials: Understanding nano-bio effects on cell behaviors. Drug Deliv. 2017, 24, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.-H.; Chang, L.W.; Lin, P. Metal-Based Nanoparticles and the Immune System: Activation, Inflammation, and Potential Applications. BioMed Res. Int. 2015, 2015, 143720. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B. Hide and Seek: Nanomaterial Interactions with the Immune System. Front. Immunol. 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chugh, H.; Sood, D.; Chandra, I.; Tomar, V.; Dhawan, G.; Chandra, R. Role of gold and silver nanoparticles in cancer nano-medicine. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.J.; Hsu, S.H.; Tsai, C.L. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small 2009, 5, 1553–1561. [Google Scholar] [CrossRef]
- Wolf-Grosse, S.; Mollnes, T.E.; Ali, S.; Stenvik, J.; Nilsen, A.M. Iron oxide nanoparticles enhance Toll-like receptor-induced cytokines in a particle size- and actin-dependent manner in human blood. Nanomedicine 2018, 13, 1773–1785. [Google Scholar] [CrossRef]
- Vasilichin, V.A.; Tsymbal, S.A.; Fakhardo, A.F.; Anastasova, E.I.; Marchenko, A.S.; Shtil, A.A.; Vinogradov, V.V.; Koshel, E.I. Effects of Metal Oxide Nanoparticles on Toll-Like Receptor mRNAs in Human Monocytes. Nanomaterials 2020, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Dykman, L.A.; Khlebtsov, N.G. Immunological properties of gold nanoparticles. Chem. Sci. 2017, 8, 1719–1735. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Park, W.; Yi, D.K. Immunostimulatory effects of gold nanorod and silica-coated gold nanorod on RAW 264.7 mouse macrophages. Toxicol. Lett. 2012, 209, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Fallarini, S.; Paoletti, T.; Battaglini, C.O.; Ronchi, P.; Lay, L.; Bonomi, R.; Jha, S.; Mancin, F.; Scrimin, P.; Lombardi, G. Factors affecting T cell responses induced by fully synthetic glyco-gold-nanoparticles. Nanoscale 2013, 5, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.Y.; Mattos Almeida, J.P.; Bear, A.; Liu, N.; Luo, L.; Foster, A.E.; Drezek, R.A. Gold Nanoparticle Delivery of Modified CpG Stimulates Macrophages and Inhibits Tumor Growth for Enhanced Immunotherapy. PLoS ONE 2013, 8, e63550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Lee, I.H.; Kang, S.; Kim, D.; Choi, M.; Saw, P.E.; Shin, E.C.; Jon, S. Gold nanoparticles displaying tumor-associated self-antigens as a potential vaccine for cancer immunotherapy. Adv. Healthc. Mater. 2014, 3, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Ninan, N.; Goswami, N.; Vasilev, K. The Impact of Engineered Silver Nanomaterials on the Immune System. Nanomaterials 2020, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Elsabahy, M.; Wooley, K.L. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem. Soc. Rev. 2013, 42, 5552–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.; Hussain, S.M.; Schrand, A.M.; Braydich-Stolle, L.K.; Hess, K.L.; Jones, R.L.; Schlager, J.J. Unique cellular interaction of silver nanoparticles: Size-dependent generation of reactive oxygen species. J. Phys. Chem. B 2008, 112, 13608–13619. [Google Scholar] [CrossRef]
- Asgary, V.; Shoari, A.; Baghbani-Arani, F.; Sadat Shandiz, S.A.; Khosravy, M.S.; Janani, A.; Bigdeli, R.; Bashar, R.; Cohan, R.A. Green synthesis and evaluation of silver nanoparticles as adjuvant in rabies veterinary vaccine. Int. J. Nanomed. 2016, 11, 3597–3605. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Balachandran, Y.L.; Li, D.; Shao, Y.; Jiang, X. Polyvinylpyrrolidone–Poly(ethylene glycol) Modified Silver Nanorods Can Be a Safe, Noncarrier Adjuvant for HIV Vaccine. ACS Nano 2016, 10, 3589–3596. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, H.; Liu, J.-H.; Wang, H.; Liu, Y. Evaluation of the adjuvant effect of silver nanoparticles both in vitro and in vivo. Toxicol. Lett. 2013, 219, 42–48. [Google Scholar] [CrossRef]
- Kim, J.V.; Latouche, J.-B.; Rivière, I.; Sadelain, M. The ABCs of artificial antigen presentation. Nat. Biotechnol. 2004, 22, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, K.R.; Green, J.J. Nanoscale artificial antigen presenting cells for cancer immunotherapy. Mol. Immunol. 2018, 98, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Eksteen-Akeroyd, Z.H.; Jacobs, M.J.; Hammink, R.; Koepf, M.; Lambeck, A.J.A.; van Hest, J.C.M.; Wilson, C.J.; Blank, K.; Figdor, C.G.; et al. Therapeutic nanoworms: Towards novel synthetic dendritic cells for immunotherapy. Chem. Sci. 2013, 4, 4168–4174. [Google Scholar] [CrossRef] [Green Version]
- Kosmides, A.K.; Meyer, R.A.; Hickey, J.W.; Aje, K.; Cheung, K.N.; Green, J.J.; Schneck, J.P. Biomimetic biodegradable artificial antigen presenting cells synergize with PD-1 blockade to treat melanoma. Biomaterials 2017, 118, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Jiang, F.; Lv, X.; Zhang, R.; Lu, A.; Zhang, G. A Mini-Review for Cancer Immunotherapy: Molecular Understanding of PD-1/PD-L1 Pathway & Translational Blockade of Immune Checkpoints. Int. J. Mol. Sci. 2016, 17, 1151. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int. Immunopharmacol. 2017, 46, 210–219. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Ma, J.; Han, W.; Wang, X.; Fang, Y.; Li, D.; Pan, H.; Zhang, L. The anticancer immune response of anti-PD-1/PD-L1 and the genetic determinants of response to anti-PD-1/PD-L1 antibodies in cancer patients. Oncotarget 2015, 6, 19393–19404. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spísek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Syed, Y.Y. Atezolizumab (in Combination with Nab-Paclitaxel): A Review in Advanced Triple-Negative Breast Cancer. Drugs 2020, 80, 601–607. [Google Scholar] [CrossRef]
- Bonvalot, S.; Rutkowski, P.L.; Thariat, J.; Carrère, S.; Ducassou, A.; Sunyach, M.P.; Agoston, P.; Hong, A.; Mervoyer, A.; Rastrelli, M.; et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): A multicentre, phase 2-3, randomised, controlled trial. Lancet Oncol. 2019, 20, 1148–1159. [Google Scholar] [CrossRef]
- Bonvalot, S.; Le Pechoux, C.; De Baere, T.; Kantor, G.; Buy, X.; Stoeckle, E.; Terrier, P.; Sargos, P.; Coindre, J.M.; Lassau, N.; et al. First-in-Human Study Testing a New Radioenhancer Using Nanoparticles (NBTXR3) Activated by Radiation Therapy in Patients with Locally Advanced Soft Tissue Sarcomas. Clin. Cancer Res. 2017, 23, 908–917. [Google Scholar] [CrossRef] [Green Version]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Dychter, S.S.; Devgan, G.; Monk, B.J. Avelumab (anti-PD-L1) in platinum-resistant/refractory ovarian cancer: JAVELIN Ovarian 200 Phase III study design. Future Oncol. 2018, 14, 2103–2113. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Clinical Translation of Nanomedicine and Biomaterials for Cancer Immunotherapy: Progress and Perspectives. Adv. Ther. 2020, 3, 1900215. [Google Scholar] [CrossRef]
- Mikelez-Alonso, I.; Aires, A.; Cortajarena, A.L. Cancer Nano-Immunotherapy from the Injection to the Target: The Role of Protein Corona. Int. J. Mol. Sci. 2020, 21, 519. [Google Scholar] [CrossRef] [Green Version]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., III; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase 1, open-label, multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in subjects with resected solid tumors and in combination with pembrolizumab in subjects with unresectable solid tumors (Keynote-603). J. Glob. Oncol. 2019, 5, 93. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Pfannenstiel, L.W.; Lam, S.S.; Emens, L.A.; Jaffee, E.M.; Armstrong, T.D. Paclitaxel enhances early dendritic cell maturation and function through TLR4 signaling in mice. Cell. Immunol. 2010, 263, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Graziani, S.R.; Vital, C.G.; Morikawa, A.T.; Van Eyll, B.M.; Fernandes Junior, H.J.; Kalil Filho, R.; Maranhão, R.C. Phase II study of paclitaxel associated with lipid core nanoparticles (LDE) as third-line treatment of patients with epithelial ovarian carcinoma. Med. Oncol. 2017, 34, 151. [Google Scholar] [CrossRef]
- Shen, C.; Frakes, J.; Weiss, J.; Caudell, J.J.; Hackman, T.G.; Akulian, J.A.; El-Haddad, G.; Hu, Y.; Dixon, R.; Pearson, A.T.; et al. Phase I study of NBTXR3 activated by radiotherapy in patients with advanced cancers treated with an anti-PD-1 therapy. J. Clin. Oncol. 2020, 38, TPS3173. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Formulation Description | Mechanism of Action | Clinical Trials | Approved by the FDA | Ref |
|---|---|---|---|---|---|
| RNA-LPX (Lipoplex®) | RNA-lipoplexes | DC maturation, T cell response | Phase I (2016) | [82] | |
| MRX34 | miRNA-34a-loaded liposome | Downregulation of immune evasion tumor genes | Phase I (2016) | [183] | |
| mRNA-4157 | mRNA-4157 encapsulated in Lipids | induce neoantigen specific T cells and associated anti-tumor responses. | Phase I (2019) | [184] | |
| Ferumoxytol (Ferahem®) | Iron oxide nanoparticles (IONP) | M2 Macrophage polarization to M1-like | Yes, for anemia and kidney diseases | [185] | |
| PTX-LDE | Paclitaxel-loaded lipid core NPs | DC maturation | Phase II (2017) | [186,187] | |
| Anti-EGFR-IL-dox | Doxorubicin-loaded anti-EGFR immunoliposomes | Block EGFR-mediated growth signaling and induce immunogenic cell death | Phase II (2016) | NCT02833766 | |
| JVRS-100 | Cationic liposome incorporating plasmid DNA complex | Immune system stimulation | Phase I (2016) | NCT00860522 | |
| NBTXR3 | Hafnium oxide nanoparticles in combination with anti-PD1 | Enhance tumor cell death via electron production, induce immunogenic cell death leading to activation of the immune system | Phase I (2019) | [188], NCT03589339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debele, T.A.; Yeh, C.-F.; Su, W.-P. Cancer Immunotherapy and Application of Nanoparticles in Cancers Immunotherapy as the Delivery of Immunotherapeutic Agents and as the Immunomodulators. Cancers 2020, 12, 3773. https://doi.org/10.3390/cancers12123773
Debele TA, Yeh C-F, Su W-P. Cancer Immunotherapy and Application of Nanoparticles in Cancers Immunotherapy as the Delivery of Immunotherapeutic Agents and as the Immunomodulators. Cancers. 2020; 12(12):3773. https://doi.org/10.3390/cancers12123773
Chicago/Turabian StyleDebele, Tilahun Ayane, Cheng-Fa Yeh, and Wen-Pin Su. 2020. "Cancer Immunotherapy and Application of Nanoparticles in Cancers Immunotherapy as the Delivery of Immunotherapeutic Agents and as the Immunomodulators" Cancers 12, no. 12: 3773. https://doi.org/10.3390/cancers12123773