Exploiting GRK2 Inhibition as a Therapeutic Option in Experimental Cancer Treatment: Role of p53-Induced Mitochondrial Apoptosis


 , ,
, ,  , and
, and
Abstract
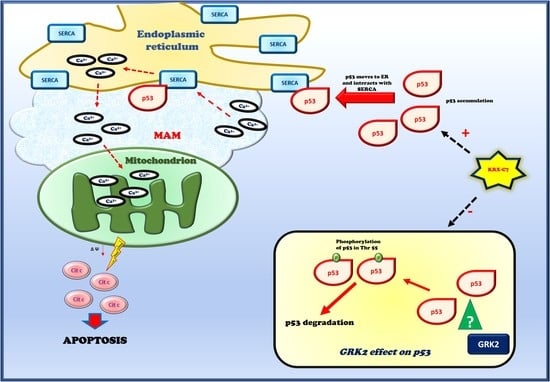
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Cell Culture
2.2. GRK2 Overexpression and Silencing
2.3. Endoplasmic Reticulum Extracts
2.4. Immunoprecipitation and Western Blotting
2.5. Immunoprecipitation and Nano-LC-MS/MS Analysis
2.6. Measurement of Mitochondrial Calcium Content
2.7. Proliferation Assay
2.8. In Vivo Study
2.9. Statistical Analysis
3. Results
3.1. GRK2 Induces Cancer Cell Proliferation by Regulating p53
3.2. GRK2 Inhibits p53 Signaling through Its Catalytic Activity
3.3. GRK2 Regulates p53 Levels by Interfering with Its Degradation
3.4. KRX-C7 Induces p53 Mitochondrial Pathway of Apoptosis
3.5. KRX-C7 Inhibits Tumor Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The Role of G Protein-coupled Receptor Kinases in Cancer. Int. J. Biol. Sci. 2018, 14, 189–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, J.; Franco, A.; Giudice, C.D.; Fiordelisi, A.; Cipolletta, E.; Ciccarelli, M.; Trimarco, B.; Iaccarino, G.; Sorriento, D. Dual role of GRK5 in cancer development and progression. Transl. Med. UniSa 2016, 14, 28–37. [Google Scholar] [PubMed]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, V.; Nogues, L.; Reglero, C.; Mayor, F., Jr.; Penela, P. Role of G protein-coupled receptor kinase 2 in tumoral angiogenesis. Mol. Cell. Oncol. 2014, 1, e969166. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Nogues, L.; Mayor, F., Jr. Role of G protein-coupled receptor kinases in cell migration. Curr. Opin. Cell Biol. 2014, 27, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Nogues, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Ramos, P.; Mendiola, M.; Berjon, A.; Stamatakis, K.; et al. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Rivas, V.; Salcedo, A.; Mayor, F., Jr. G protein-coupled receptor kinase 2 (GRK2) modulation and cell cycle progression. Proc. Natl. Acad. Sci. USA 2010, 107, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Metaye, T.; Menet, E.; Guilhot, J.; Kraimps, J.L. Expression and activity of g protein-coupled receptor kinases in differentiated thyroid carcinoma. J. Clin. Endocrinol. Metab. 2002, 87, 3279–3286. [Google Scholar] [CrossRef]
- Metaye, T.; Levillain, P.; Kraimps, J.L.; Perdrisot, R. Immunohistochemical detection, regulation and antiproliferative function of G-protein-coupled receptor kinase 2 in thyroid carcinomas. J. Endocrinol. 2008, 198, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Zaika, A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 2001, 493, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Del Giudice, C.; Bertamino, A.; Ciccarelli, M.; Gomez-Monterrey, I.; Campiglia, P.; Novellino, E.; Illario, M.; Trimarco, B.; De Luca, N.; et al. New small molecules, ISA27 and SM13, inhibit tumour growth inducing mitochondrial effects of p53. Br. J. Cancer 2015, 112, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Campanile, A.; Santulli, G.; Leggiero, E.; Pastore, L.; Trimarco, B.; Iaccarino, G. A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Mol. Cancer 2009, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, G.; Izzo, R.; Trimarco, V.; Cipolletta, E.; Lanni, F.; Sorriento, D.; Iovino, G.L.; Rozza, F.; De Luca, N.; Priante, O.; et al. Beta2-adrenergic receptor polymorphisms and treatment-induced regression of left ventricular hypertrophy in hypertension. Clin. Pharmacol. Ther. 2006, 80, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, J.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; De Rosa, M.; Sala, M.; Pacelli, R.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. A Novel Small Peptide Inhibitor of NFkappaB, RH10, Blocks Oxidative Stress-Dependent Phenotypes in Cancer. Oxid. Med. Cell Longev. 2018, 2018, 5801807. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, A.; Cipolletta, E.; Gomez-Monterrey, I.; Sala, M.; Vernieri, E.; Limatola, A.; Bertamino, A.; Musella, S.; Sorriento, D.; Grieco, P.; et al. Design, synthesis and efficacy of novel G protein-coupled receptor kinase 2 inhibitors. Eur. J. Med. Chem. 2013, 69, 384–392. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Fiordelisi, A.; Gambardella, J.; Franco, A.; Del Giudice, C.; Sala, M.; Monti, M.G.; Bertamino, A.; Campiglia, P.; et al. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020, 7, 1571–1584. [Google Scholar] [CrossRef]
- Li, H.H.; Li, A.G.; Sheppard, H.M.; Liu, X. Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: A role for TAF1 in cell G1 progression. Mol. Cell 2004, 13, 867–878. [Google Scholar] [CrossRef]
- Haupt, S.; Raghu, D.; Haupt, Y. p53 Calls upon CIA (Calcium Induced Apoptosis) to Counter Stress. Front. Oncol. 2015, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, R.; Vella, V.; Vigneri, R.; Frasca, F. p53 family proteins in thyroid cancer. Endocr. Relat. Cancer 2007, 14, 43–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamble, S.C.; Cook, M.C.; Riches, A.C.; Herceg, Z.; Bryant, P.E.; Arrand, J.E. p53 mutations in tumors derived from irradiated human thyroid epithelial cells. Mutat. Res. 1999, 425, 231–238. [Google Scholar] [CrossRef]
- Quiros, R.M.; Ding, H.G.; Gattuso, P.; Prinz, R.A.; Xu, X. Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations. Cancer 2005, 103, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Lima, J.; Preto, A.; Castro, P.; Vinagre, J.; Celestino, R.; Couto, J.P.; Prazeres, H.; Eloy, C.; Maximo, V.; et al. Genetic alterations in poorly differentiated and undifferentiated thyroid carcinomas. Curr. Genom. 2011, 12, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Donghi, R.; Longoni, A.; Pilotti, S.; Michieli, P.; Della Porta, G.; Pierotti, M.A. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Investig. 1993, 91, 1753–1760. [Google Scholar] [CrossRef]
- Sobrinho-Simoes, M.; Maximo, V.; Rocha, A.S.; Trovisco, V.; Castro, P.; Preto, A.; Lima, J.; Soares, P. Intragenic mutations in thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2008, 37, 333–362. [Google Scholar] [CrossRef]
- Benchimol, S. p53-dependent pathways of apoptosis. Cell Death Differ. 2001, 8, 1049–1051. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, H.; Yuan, M.; Fu, J.; Zhou, Y.; Ma, L. G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J. Biol. Chem. 2010, 285, 12823–12830. [Google Scholar] [CrossRef] [Green Version]
- Fusco, A.; Santulli, G.; Sorriento, D.; Cipolletta, E.; Garbi, C.; Dorn, G.W., 2nd; Trimarco, B.; Feliciello, A.; Iaccarino, G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell Signal. 2012, 24, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Fusco, A.; Ciccarelli, M.; Rungi, A.; Anastasio, A.; Carillo, A.; Dorn, G.W., 2nd; Trimarco, B.; Iaccarino, G. Mitochondrial G protein coupled receptor kinase 2 regulates proinflammatory responses in macrophages. FEBS Lett. 2013, 587, 3487–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.; Sorriento, D.; Gambardella, J.; Pacelli, R.; Prevete, N.; Procaccini, C.; Matarese, G.; Trimarco, B.; Iaccarino, G.; Ciccarelli, M. GRK2 moderates the acute mitochondrial damage to ionizing radiation exposure by promoting mitochondrial fission/fusion. Cell Death Discov. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolletta, E.; Gambardella, J.; Fiordelisi, A.; Del Giudice, C.; Di Vaia, E.; Ciccarelli, M.; Sala, M.; Campiglia, P.; Coscioni, E.; Trimarco, B.; et al. Antidiabetic and Cardioprotective Effects of Pharmacological Inhibition of GRK2 in db/db Mice. Int. J. Mol. Sci. 2019, 20, 1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Ciccarelli, M.; Cipolletta, E.; Trimarco, B.; Iaccarino, G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure—A Review on Available GRK2 Inhibitors. Front. Cardiovasc. Med. 2016, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Sorriento, D.; Santulli, G.; Franco, A.; Cipolletta, E.; Napolitano, L.; Gambardella, J.; Gomez-Monterrey, I.; Campiglia, P.; Trimarco, B.; Iaccarino, G.; et al. Integrating GRK2 and NFkappaB in the Pathophysiology of Cardiac Hypertrophy. J. Cardiovasc. Transl. Res. 2015, 8, 493–502. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; Sabra, M.M.; Brenner, A.; Moore, S.C.; Ron, E.; Schatzkin, A.; Hollenbeck, A.; Ward, M.H. Diabetes and thyroid cancer risk in the National Institutes of Health-AARP Diet and Health Study. Thyroid 2011, 21, 957–963. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambardella, J.; Fiordelisi, A.; Santulli, G.; Ciccarelli, M.; Cerasuolo, F.A.; Sala, M.; Sommella, E.; Campiglia, P.; Illario, M.; Iaccarino, G.; et al. Exploiting GRK2 Inhibition as a Therapeutic Option in Experimental Cancer Treatment: Role of p53-Induced Mitochondrial Apoptosis. Cancers 2020, 12, 3530. https://doi.org/10.3390/cancers12123530
Gambardella J, Fiordelisi A, Santulli G, Ciccarelli M, Cerasuolo FA, Sala M, Sommella E, Campiglia P, Illario M, Iaccarino G, et al. Exploiting GRK2 Inhibition as a Therapeutic Option in Experimental Cancer Treatment: Role of p53-Induced Mitochondrial Apoptosis. Cancers. 2020; 12(12):3530. https://doi.org/10.3390/cancers12123530
Chicago/Turabian StyleGambardella, Jessica, Antonella Fiordelisi, Gaetano Santulli, Michele Ciccarelli, Federica Andrea Cerasuolo, Marina Sala, Eduardo Sommella, Pietro Campiglia, Maddalena Illario, Guido Iaccarino, and et al. 2020. "Exploiting GRK2 Inhibition as a Therapeutic Option in Experimental Cancer Treatment: Role of p53-Induced Mitochondrial Apoptosis" Cancers 12, no. 12: 3530. https://doi.org/10.3390/cancers12123530