Making Connections: p53 and the Cathepsin Proteases as Co-Regulators of Cancer and Apoptosis


Abstract
:Simple Summary
Abstract
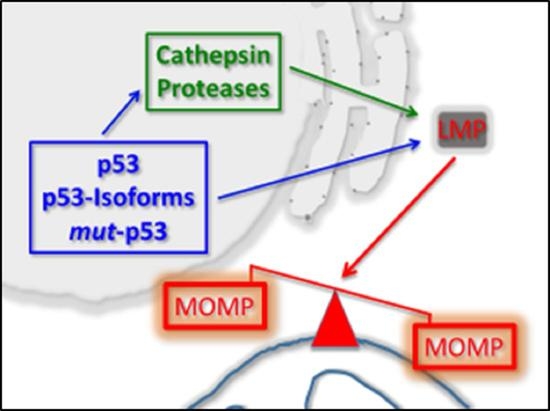
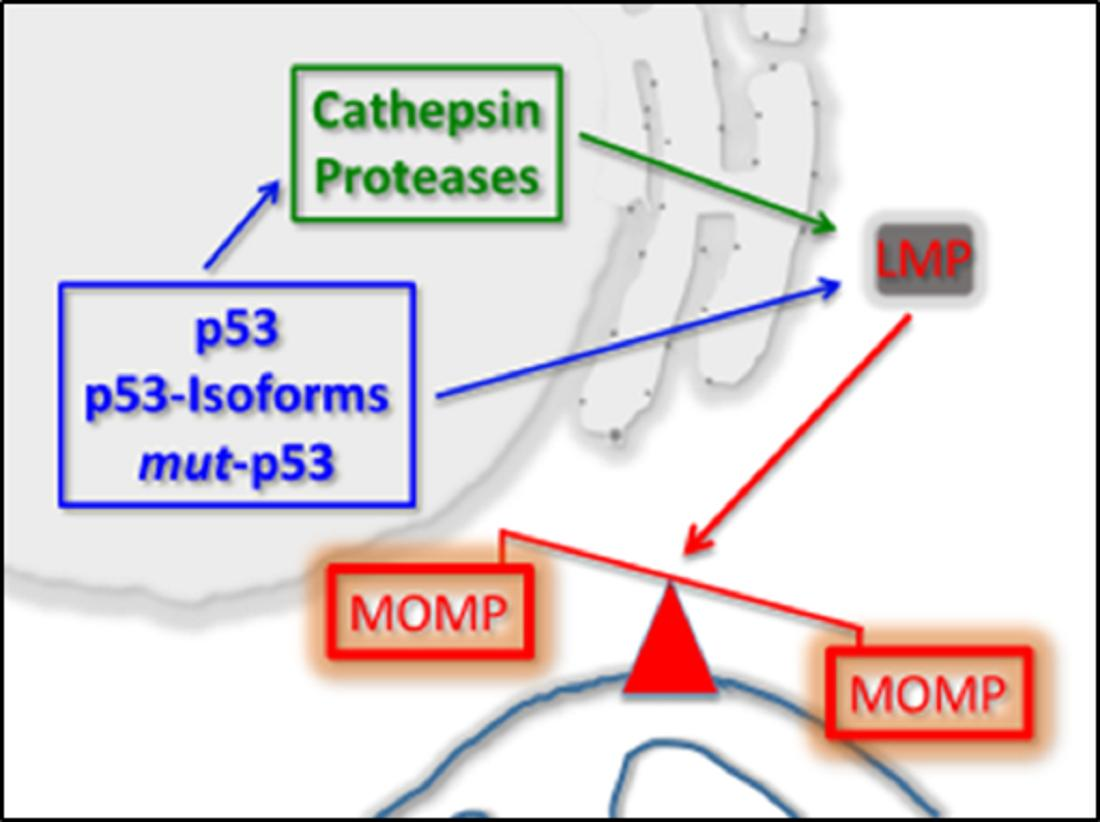
1. Introduction
1.1. p53 Transcription and Functionality: An Update
1.2. Lysosomal Membrane Permeabilization—A Key Event in Cell Death
1.3. p53 As a Co-Regulator of Mitochondrial and Lysosomal Mediated Cell Death
1.4. p53 Activation and Cathepsin Protein Expression
1.5. p53 and Cathepsin Expression in Tumor Samples: A Clinically Relevant Relationship
1.6. Cathepsin Genes As Potential Targets for p53
2. Future Directions
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nat. Cell Biol. 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, 026070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.-A.T.; Menendez, D.; A Resnick, M.; Anderson, C.W. Mutant TP53 posttranslational modifications: Challenges and opportunities. Hum. Mutat. 2014, 35, 738–755. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Molchadsky, A.; Rotter, V. p53 and its mutants on the slippery road from stemness to carcinogenesis. Carcinogenesis 2017, 38, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Pfeifer, G.P. SomaticTP53Mutations in the era of genome sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, 026179. [Google Scholar] [CrossRef] [Green Version]
- Gencel-Augusto, J.; Lozano, G. p53 tetramerization: At the center of the dominant-negative effect of mutant p53. Genes Dev. 2020, 34, 1128–1146. [Google Scholar] [CrossRef]
- McLure, K.G. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef]
- Jimenez, G.S.; Khan, S.H.; Stommel, J.M.; Wahl, G.M. p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 1999, 18, 7656–7665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, 000950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Levine, A.J. Differential regulation of the p21/WAF-1 and mdm2 genes after high-dose UV irradiation: p53-dependent and p53-independent regulation of the mdm2 gene. Mol. Med. 1997, 3, 441–451. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar]
- Lohrum, M.A.E.; Woods, D.B.; Ludwig, R.L.; Balint, E.; Vousden, K.H. C-terminal ubiquitination of p53 contributes to nuclear export. Mol. Cell. Biol. 2001, 21, 8521–8532. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Stanek, T.J.; Frank, A.; Murphy, M.E.; McMahon, S.B. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J. Biol. Chem. 2009, 284, 20197–20205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, G.; Mioni, M.; Giorgi, C.; Ruggeri, N.; Pinton, P.; Moll, U.; Mantovani, F.; Del Sal, G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ. 2012, 20, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, F.; Di Conza, G.; Pellegrino, M.; Rinaldo, C.; Prodosmo, A.; Giglio, S.; D’Agnano, I.; Florenzano, F.; Felicioni, L.; Buttitta, F.; et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J. 2009, 28, 1926–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; A Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, F.; Moretti, F. Mitochondrial MDM4 (MDMX): An unpredicted role in the p53-mediated intrinsic apoptotic pathway. Cell Cycle 2009, 8, 3854–3859. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B.; et al. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, 000935. [Google Scholar] [CrossRef] [Green Version]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, 026104. [Google Scholar] [CrossRef]
- Helton, E.S.; Chen, X. p53 modulation of the DNA damage response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Murcia, J.M.D.; Susin, S.A. Sequential activation of poly(ADP-Ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haince, J.-F.; Rouleau, M.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G. Targeting poly(ADP-ribosyl)ation: A promising approach in cancer therapy. Trends Mol. Med. 2005, 11, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Shall, S.; De Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. Repair 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Fortin, A.; Cregan, S.P.; Maclaurin, J.G.; Kushwaha, N.; Hickman, E.S.; Thompson, C.S.; Hakim, A.; Albert, P.R.; Cecconi, F.; Helin, K.; et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J. Cell Biol. 2001, 155, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroni, M.C.; Hickman, E.S.; Denchi, E.L.; Caprara, G.; Colli, E.; Cecconi, F.; Müller, H.; Helin, K. Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Thornborrow, E.C.; Patel, S.; Mastropietro, A.E.; Schwartzfarb, E.M.; Manfredi, J.J. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene 2002, 21, 990–999. [Google Scholar] [CrossRef] [Green Version]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Happo, L.; Cragg, M.S.; Phipson, B.; Haga, J.M.; Jansen, E.S.; Herold, M.J.; Dewson, G.; Michalak, E.M.; Vandenberg, C.J.; Smyth, G.K.; et al. Maximal killing of lymphoma cells by DNA damage–inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 2010, 116, 5256–5267. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, E.; Wang, Z.; Kelly, G.L.; Strasser, A. Discussion of some ‘knowns’ and some ‘unknowns’ about the tumour suppressor p53. J. Mol. Cell Biol. 2019, 11, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Budhram-Mahadeo, V.; Morris, P.J.; Smith, M.D.; Midgley, C.A.; Boxer, L.M.; Latchman, D.S. p53 suppresses the activation of the Bcl-2 promoter by the Brn-3a POU family transcription factor. J. Biol. Chem. 1999, 274, 15237–15244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommer, G.T.; Gerin, I.; Feng, Y.; Kaczorowski, A.J.; Kuick, R.; Love, R.E.; Zhai, Y.; Giordano, T.J.; Qin, Z.S.; Moore, B.B.; et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007, 17, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; A Liebermann, D.; Hoffman, B.; Reed, J.C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Sugars, K.L.; Budhram-Mahadeo, V.; Packham, G.; Latchman, D.S. A minimal Bcl-x promoter is activated by Brn-3a and repressed by p53. Nucleic Acids Res. 2001, 29, 4530–4540. [Google Scholar] [CrossRef] [Green Version]
- Pietrzak, M.; Puzianowska-Kuznicka, M. p53-dependent repression of the human MCL-1 gene encoding an anti-apoptotic member of the BCL-2 family: The role of Sp1 and of basic transcription factor binding sites in the MCL-1 promoter. Biol. Chem. 2008, 389, 383–393. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Sinkovic, T.; Combs, S.E.; Roth, W. p53-dependent regulation of Mcl-1 contributes to synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL inhibitor ABT-737. Apoptosis 2012, 17, 187–199. [Google Scholar] [CrossRef]
- Liu, J.; Chen, G.; Feng, L.; Zhang, W.; Pelicano, H.; Wang, F.; Ogasawara, M.A.; Lu, W.; Amin, H.M.; Croce, C.M.; et al. Loss of p53 and altered miR15-a/16-1short right arrowMCL-1 pathway in CLL: Insights from TCL1-Tg:p53(-/-) mouse model and primary human leukemia cells. Leukemia 2014, 28, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nat. Cell Biol. 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nat. Cell Biol. 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Van Loo, G.; Schotte, P.; Van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Genet. 2002, 9, 680–684. [Google Scholar] [CrossRef]
- Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP-1: Friend or foe of DNA damage and repair in tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Zamyatnin, A.A., Jr. ‘Patchiness’ and basic cancer research: Unravelling the proteases. Cell Cycle 2019, 18, 1687–1701. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, J.A.A. Cysteine Cathepsin protease inhibition: An update on its diagnostic, prognostic and therapeutic potential in cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Soond, S.M.; Kozhevnikova, M.V.; Frolova, A.S.; Savvateeva, L.V.; Plotnikov, E.Y.; Townsend, P.A.; Han, Y.P.; Zamyatnin, A.A. Lost or forgotten: The nuclear cathepsin protein isoforms in cancer. Cancer Lett. 2019, 462, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Johansson, A.C.; Linderoth, E.; Johansson, U.; Antonsson, B.; Steinfeld, R.; Kågedal, K.; Öllinger, K. Lysosome-mediated apoptosis is associated with cathepsin D-specific processing of bid at Phe24, Trp48, and Phe183. Ann. Clin. Lab. Sci. 2012, 42, 231–242. [Google Scholar] [PubMed]
- Cirman, T.; Orešić, K.; Mazovec, G.D.; Turk, V.; Reed, J.C.; Myers, R.M.; Salvesen, G.S.; Turk, B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2003, 279, 3578–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droga-Mazovec, G.; Bojič, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine Cathepsins trigger caspase-dependent cell death through cleavage of Bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Ohsakaya, S.; Nagaura, Z.-I.; Ishihara, N.; Mihara, K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005, 24, 1375–1386. [Google Scholar] [CrossRef]
- Polster, B.M.; Basañez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2004, 280, 6447–6454. [Google Scholar] [CrossRef] [Green Version]
- Yuste, V.J.; Moubarak, R.S.; Delettre, C.; Bras, M.; Sancho, P.; Robert, N.; D’Alayer, J.; Susin, S.A. Cysteine protease inhibition prevents mitochondrial apoptosis-inducing factor (AIF) release. Cell Death Differ. 2005, 12, 1445–1448. [Google Scholar] [CrossRef]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ikeguchi, M.; Sakatani, T.; Ueta, T.; Fukuda, K.; Oka, S.; Hisamitsu, K.; Yamaguchi, K.; Tsujitani, S.; Kaibara, N. Correlation between cathepsin D expression and p53 protein nuclear accumulation in oesophageal squamous cell carcinoma. J. Clin. Pathol. 2002, 55, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, H.; Babic, R.; Grützner, K.U.; Beyer, B.C.; Tarabichi, A.; Schildberg, F.W.; Heiss, M.M. An immunohistochemical assessment of cathepsin D in gastric carcinoma: Its impact on clinical prognosis. Cancer 1997, 80, 179–187. [Google Scholar] [CrossRef]
- Vigneswaran, N.; Zhao, W.; Dassanayake, A.; Muller, S.; Miller, N.M.; Zacharias, W. Variable expression of cathepsin B and D correlates with highly invasive and metastatic phenotype of oral cancer. Hum. Pathol. 2000, 31, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Adenis, A.; Huet, G.; Zerimech, F.; Hecquet, B.; Balduyck, M.; Peyrat, J. Cathepsin B, L, and D activities in colorectal carcinomas: Relationship with clinico-pathological parameters. Cancer Lett. 1995, 96, 267–275. [Google Scholar] [CrossRef]
- Kang, G.H.; Lee, B.S.; Lee, E.S.; Kim, S.H.; Lee, H.Y.; Kang, D.Y. Prognostic Significance of p53, mTOR, c-Met, IGF-1R, and HSP70 overexpression after the resection of hepatocellular carcinoma. Gut Liver 2014, 8, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, 026146. [Google Scholar] [CrossRef] [Green Version]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Kashiyama, T.; Oda, K.; Ikeda, Y.; Shiose, Y.; Hirota, Y.; Inaba, K.; Makii, C.; Kurikawa, R.; Miyasaka, A.; Koso, T.; et al. Antitumor Activity and Induction of TP53-Dependent Apoptosis toward Ovarian Clear Cell Adenocarcinoma by the Dual PI3K/mTOR Inhibitor DS-7423. PLoS ONE 2014, 9, 87220. [Google Scholar] [CrossRef] [Green Version]
- Lunova, M.; Smolková, B.; Lynnyk, A.; Uzhytchak, M.; Jirsa, M.; Kubinova, S.; Dejneka, A.; Lunov, O. Targeting the mTOR signaling pathway utilizing nanoparticles: A critical overview. Cancers 2019, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K. p53 isoforms in cellular senescence-and ageing-associated biological and physiological functions. Int. J. Mol. Sci. 2019, 20, 6023. [Google Scholar] [CrossRef] [Green Version]
- Anbarasan, T.; Bourdon, J.C. The emerging landscape of p53 isoforms in physiology, cancer and degenerative diseases. Int. J. Mol. Sci. 2019, 20, 6257. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.J.; Gaiddon, C.; Storr, T. A balancing act: Using small molecules for therapeutic intervention of the p53 pathway in cancer. Chem. Soc. Rev. 2020, 49, 6995–7014. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; A Bonneau, R.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Loh, S.N. The missing Zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; Dedecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Wilcken, R.; Liu, X.; Zimmermann, M.O.; Rutherford, T.J.; Fersht, A.R.; Joerger, A.C.; Boeckler, F.M. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J. Am. Chem. Soc. 2012, 134, 6810–6818. [Google Scholar] [CrossRef]
- Silva, J.L.; Cino, E.A.; Soares, I.N.; Ferreira, V.F.; De Oliveira, G.A.P. Targeting the prion-like aggregation of mutant p53 to combat cancer. Accounts Chem. Res. 2017, 51, 181–190. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 amyloids in cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA control of p53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, J.; Etemadi, T.; Alyasin, A. The P53/microRNA network: A potential tumor suppressor with a role in anticancer therapy. Pharmacol. Res. 2020, 160, 105179. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.H.; Chao, C.F.; Huang, H.C.; Lee, H.Y.; Kannagi, R.; Chen, J.Y. Roles of p53 family structure and function in non-canonical response element binding and activation. Int. J. Mol. Sci. 2019, 20, 3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazda, V.; Fojta, M. The rich world of p53 DNA binding targets: The role of DNA structure. Int. J. Mol. Sci. 2019, 20, 5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [Green Version]
- Stindt, M.H.; Muller, P.A.J.; Ludwig, R.L.; Kehrloesser, S.; Dotsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2014, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Kim, S.O.; Han, J. Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol. Cell. Biol. 2003, 23, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Firestone, R.A.; Pisano, J.M.; Bonney, R.J. Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J. Med. Chem. 1979, 22, 1130–1133. [Google Scholar] [CrossRef]
- Kagedal, K.; Zhao, M.; Svensson, I.; Brunk, U.T. Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 2001, 359, 335–343. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, H.; Hu, Z.; Zhu, M.; Wang, L.; Han, L.; Qian, L.; Tian, K.; Yuan, H.; Lou, H.; et al. Targeting the lysosome by an aminomethylated Riccardin D triggers DNA damage through cathepsin B-mediated degradation of BRCA1. J. Cell. Mol. Med. 2018, 23, 1798–1812. [Google Scholar] [CrossRef]
- Ostenfeld, M.S.; Fehrenbacher, N.; Hoyer-Hansen, M.; Thomsen, C.; Farkas, T.; Jaattela, M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005, 65, 8975–8983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostenfeld, M.S.; Høyer-Hansen, M.; Bastholm, L.; Fehrenbacher, N.; Olsen, O.D.; Groth-Pedersen, L.; Puustinen, P.; Kirkegaard-Sørensen, T.; Nylandsted, J.; Farkas, T.; et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008, 4, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, R.; Dokic, I.; Kees, T.; Hüber, C.M.; Keitel, D.; Geibig, R.; Brügge, B.; Zentgraf, H.; Brady, N.R.; Régnier-Vigouroux, A.; et al. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364–1383. [Google Scholar] [CrossRef] [PubMed]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullio, C.; Casas, J.; Brunk, U.T.; Sala, G.; Fabrias, G.; Ghidoni, R.; Bonelli, G.; Baccino, F.M.; Autelli, R. Sphingosine mediates TNFα-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J. Lipid Res. 2012, 53, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schütze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J. 2010, 30, 379–394. [Google Scholar] [CrossRef]
- Liu, N.; Raja, S.M.; Zazzeroni, F.; Metkar, S.S.; Shah, R.; Zhang, M.; Wang, Y.; Brömme, D.; Russin, W.A.; Lee, J.C.; et al. Ashton-Rickardt, NF-kappaB protects from the lysosomal pathway of cell death. EMBO J. 2003, 22, 5313–5322. [Google Scholar] [CrossRef] [Green Version]
- Taha, T.A.; Kitatani, K.; Bielawski, J.; Cho, W.; Hannun, Y.A.; Obeid, L.M. Tumor necrosis factor induces the loss of sphingosine kinase-1 by a cathepsin B-dependent mechanism. J. Biol. Chem. 2005, 280, 17196–17202. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Yu, C.J.; Luh, K.T.; Kuo, S.H.; Lee, Y.C.; Yang, P.C. Aberrant p53 expression correlates with expression of vascular endothelial growth factor mRNA and Interleukin-8 mRNA and Neoangiogenesis in non–SMALL-cell lung cancer. J. Clin. Oncol. 2002, 20, 900–910. [Google Scholar]
- Fehrenbacher, N.; Bastholm, L.; Kirkegaard-Sørensen, T.; Rafn, B.; Bøttzauw, T.; Nielsen, C.; Weber, E.; Shirasawa, S.; Kallunki, T.; Jäättelä, M.; et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008, 68, 6623–6633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mareninova, O.A.; Sendler, M.; Malla, S.R.; Yakubov, I.; French, S.W.; Tokhtaeva, E.; Vagin, O.; Oorschot, V.; Lüllmann-Rauch, R.; Blanz, J.; et al. Lysosome-associated membrane proteins (LAMP) maintain pancreatic acinar cell homeostasis: LAMP-2–deficient mice develop pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 678–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdal, H.; Berndtsson, M.; Castro, J.; Brunk, U.; Shoshan, M.C.; Linder, S. Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, N.H.; Olsen, O.D.; Groth-Pedersen, L.; Ellegaard, A.M.; Bilgin, M.; Redmer, S.; Ostenfeld, M.S.; Ulanet, D.; Dovmark, T.H.; Lønborg, A.; et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 2013, 24, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J. Clin. Investig. 2013, 123, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Mena, S.; Rodríguez, M.L.; Ponsoda, X.; Estrela, J.M.; Jäättela, M.; Ortega, A.L. Pterostilbene-induced tumor cytotoxicity: A lysosomal membrane permeabilization-dependent mechanism. PLoS ONE 2012, 7, 44524. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Shi, J.G.; Yao, Q.H.; Jiao, D.M.; Wang, Y.Y.; Hu, H.Z.; Wu, Y.Q.; Song, J.; Yan, J.; Wu, L.J.; et al. Lysosomal membrane permeabilization is involved in curcumin-induced apoptosis of A549 lung carcinoma cells. Mol. Cell. Biochem. 2011, 359, 389–398. [Google Scholar] [CrossRef]
- Mijatovic, T.; Mathieu, V.; Gaussin, J.F.; De Nève, N.; Ribaucour, F.; Van Quaquebeke, E.; Dumont, P.; Darro, F.; Kiss, R. Cardenolide-induced lysosomal membrane permeabilization demonstrates therapeutic benefits in experimental human non-small cell lung cancers1. Neoplasia 2006, 8, 402–412. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Perchiniak, E.; Canutescu, A.A.; Wang, G.; Dunbrack, R.L.; Murphy, M.E. Oligomerization of BAK by p53 Utilizes Conserved Residues of the p53 DNA Binding Domain. J. Biol. Chem. 2008, 283, 21294–21304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, J.I.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell. Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.M.; Li, W.; Dalen, H.; Lotem, J.; Kama, R.; Sachs, L.; Brunk, U.T. Lysosomal destabilization in p53-induced apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 6286–6291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zheng, Y.; Chen, W.; Wang, C.; Liu, X.; He, W.; Xu, H.; Cao, X. Adaptor protein LAPF recruits phosphorylated p53 to lysosomes and triggers lysosomal destabilization in apoptosis. Cancer Res. 2007, 67, 11176–11185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Kasugai, T.; Ishiguro, S. Influence of cathepsin D expression in lung adenocarcinoma on prognosis: Possible importance of its expression in tumor cells and stromal cells, and its intracellular polarization in tumor cells. J. Surg. Oncol. 1997, 65, 10–19. [Google Scholar] [CrossRef]
- Sloman, A.; D’Amico, F.; A Yousem, S. Immunohistochemical markers of prolonged survival in small cell carcinoma of the lung. An immunohistochemical study. Arch. Pathol. Lab. Med. 1996, 120, 465–472. [Google Scholar]
- Ahrendt, S.A.; Chow, J.T.; Xu, L.-H.; Yang, S.C.; Eisenberger, C.F.; Esteller, M.; Herman, J.G.; Wu, L.; Decker, P.A.; Jen, J.; et al. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J. Natl. Cancer Inst. 1999, 91, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Lazaris, A.C.; Lendari, I.; Kavantzas, N.; Kandiloros, D.; Adamopoulos, G.; Davaris, P. Correlation of tumor markers p53, bcl-2 and cathepsin-D with clinicopathologic features and disease-free survival in laryngeal squamous cell carcinoma. Pathol. Int. 2000, 50, 717–724. [Google Scholar] [CrossRef]
- Charpin, C.; Bonnier, P.; Khouzami, A.; Vacheret, H.; Andrac, L.; Lavaut, M.N.; Allasia, C.; Piana, L. Inflammatory breast carcinoma: An immunohistochemical study using monoclonal anti-pHER-2/neu, pS2, cathepsin, ER and PR. Anticancer. Res. 1992, 12, 591–597. [Google Scholar]
- Aziz, S.; Pervez, S.; Khan, S.; Siddiqui, T.; Kayani, N.; Israr, M.; Rahbar, M. Case control study of novel prognostic markers and disease outcome in pregnancy/lactation-associated breast carcinoma. Pathol. Res. Pract. 2003, 199, 15–21. [Google Scholar] [CrossRef]
- Ioachim, E.; Tsanou, E.; Briasoulis, E.; Batsis, C.; Karavasilis, V.; Charchanti, A.; Pavlidis, N.; Agnantis, N.J. Clinicopathological study of the expression of hsp27, pS2, cathepsin D and metallothionein in primary invasive breast cancer. Breast 2003, 12, 111–119. [Google Scholar] [CrossRef]
- Kanno, Y.; Takane, Y.; Izawa, T.; Nakahama, T.; Inouye, Y. The inhibitory effect of aryl hydrocarbon receptor repressor (AhRR) on the growth of human breast cancer MCF-7 Cells. Biol. Pharm. Bull. 2006, 29, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lösch, A.; Schindl, M.; Kohlberger, P.; Lahodny, J.; Breitenecker, G.; Horvat, R.; Birner, P. Cathepsin D in ovarian cancer: Prognostic value and correlation with p53 expression and microvessel density. Gynecol. Oncol. 2004, 92, 545–552. [Google Scholar] [CrossRef]
- Shin, I.Y.; Sung, N.Y.; Lee, Y.S.; Kwon, T.S.; Si, Y.; Lee, Y.S.; Oh, S.T.; Lee, I.K. The expression of multiple proteins as prognostic factors in colorectal cancer: Cathepsin D, p53, COX-2, epidermal growth factor receptor, C-erbB-2, and Ki-67. Gut Liver 2014, 8, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Guerra, E.; Cimadamore, A.; Simeone, P.; Vacca, G.; Lattanzio, R.; Botti, G.; Gatta, V.; D’Aurora, M.; Simionati, B.; Piantelli, M.; et al. p53, cathepsin D, Bcl-2 are joint prognostic indicators of breast cancer metastatic spreading. BMC Cancer 2016, 16, 649. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Kurosaka, Y.; Fushida, S.; Kanno, M.; Yonemura, Y.; Miwa, K.; Miyazaki, I. Expression of p53 protein in colorectal cancer and its relationship to short-term prognosis. Cancer 1992, 70, 2778–2784. [Google Scholar] [CrossRef] [Green Version]
- Soong, R.; Grieu, F.; Robbins, P.; Dix, B.; Chen, D.; Parsons, R.; House, A.; Iacopetta, B. p53 alterations are associated with improved prognosis in distal colonic carcinomas. Clin. Cancer Res. 1997, 3, 1405–1411. [Google Scholar]
- Smith, D.R.; Ji, C.Y.; Goh, H.S. Prognostic significance of p53 overexpression and mutation in colorectal adenocarcinomas. Br. J. Cancer 1996, 74, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Aziz, S.A.; Pervez, S.; Khan, S.; Kayani, N.; Azam, S.I.; Rahbar, M.H. Case control study of prognostic markers and disease outcome in inflammatory carcinoma breast: A unique clinical experience. Breast J. 2001, 7, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, D.; Krysiak, O.; Inga, A.; Krysiak, B.; A Resnick, M.; Schönfelder, G. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc. Natl. Acad. Sci. USA 2006, 103, 1406–1411. [Google Scholar] [CrossRef] [Green Version]
- Menendez, D.; Inga, A.; Snipe, J.; Krysiak, O.; Schönfelder, G.; A Resnick, M. A Single-nucleotide polymorphism in a half-binding site creates p53 and estrogen receptor control of vascular endothelial growth factor receptor 1. Mol. Cell. Biol. 2007, 27, 2590–2600. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.J.; Menendez, D.; Inga, A.; Noureddine, M.; Bell, D.A.; Resnick, M.A. Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet. 2008, 4, 1000104. [Google Scholar] [CrossRef]
- Wu, G.S.; Saftig, P.; Peters, C.; El-Deiry, W.S. Potential role for Cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 1998, 16, 2177–2183. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.G.; Lopes, C.F.B.; Júnior, C.G.C.; Thomé, R.G.; Dos Santos, H.B.; Reis, R.M.V.; de Azambuja Ribeiro, R.I.M. WIN55,212-2 induces caspase-independent apoptosis on human glioblastoma cells by regulating HSP70, p53 and Cathepsin D. Toxicol. Vitr. 2019, 57, 233–243. [Google Scholar] [CrossRef]
- Katara, R.; Mir, R.A.; Shukla, A.A.; Tiwari, A.; Singh, N.; Chauhan, S.S. Wild type p53-dependent transcriptional upregulation of cathepsin L expression is mediated by C/EBPα03B1; in human glioblastoma cells. Biol. Chem. 2010, 391, 1031–1040. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Wang, W.J.; Li, J.; Yang, N.; Chen, G.; Wang, Z.; Liang, Z.Q. Cathepsin L suppression increases the radiosensitivity of human glioma U251 cells via G2/M cell cycle arrest and DNA damage. Acta Pharmacol. Sin. 2015, 36, 1113–1125. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xiong, Y.; Ding, X.; Wang, L.; Zhao, Y.; Fei, Y.; Zhu, Y.; Shen, X.; Tan, C.; Liang, Z.; et al. Cathepsin L activated by mutant p53 and Egr-1 promotes ionizing radiation-induced EMT in human NSCLC. J. Exp. Clin. Cancer Res. 2019, 38, 61. [Google Scholar] [CrossRef]
- Cui, D.; Wang, L.; Jiang, W.; Qi, A.; Zhou, Q.; Zhang, X. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-κB/p53 signaling pathway. Neuroscience 2013, 246, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Jin, X.; Cui, X.; Jin, C.; Piao, L.; Wan, Y.; Xu, S.; Zhang, S.; Yue, X.; Wang, H.; et al. Dipeptidyl peptidase-4 inhibition prevents vascular aging in mice under chronic stress: Modulation of oxidative stress and inflammation. Chem. Interact. 2019, 314, 108842. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-A.T.; A Grimm, S.; Bushel, P.R.; Li, J.; Li, Y.; Bennett, B.D.; A Lavender, C.; Ward, J.M.; Fargo, D.C.; Anderson, C.W.; et al. Revealing a human p53 universe. Nucleic Acids Res. 2018, 46, 8153–8167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| p53 Expression | Cathepsin | Cells | Reference |
|---|---|---|---|
| WT-p53 increased | D | Lung, Ovarian, Leukemia cells | [130] |
| mut-p53 increased | L | Glioblastoma | [132] |
| WT-p53 increased | B | Rat hippocampus | [135] |
| mut-p53 increased | L | Glioblastoma | [133] |
| WT-p53 increased | S, K | Mouse Aorta | [136] |
| WT-p53 increased | D | Glioblastoma Cell lines | [131] |
| mut-p53 increased | L | Non-small cell lung cancer | [134] |
| p53 Positive | Cathepsin Positive | Study Size | Cancer Type | Reference | Survival |
|---|---|---|---|---|---|
| +D (47%) | 152 | LAC | [137] | poor | |
| −B (23%) −D (87%) | 28 | SCCL | [138] | prolonged prolonged | |
| 57.8% | +D (31.25%) +D (37.50%) | 64 | SCC larynx | [140] | high risk of relapse |
| 70% | +D (30%) | 40 | IBC | [125] | poor |
| 46% | +D (49%) | 154 | SCC Oesphageal | [70] | Invasiveness/ poor prognosis |
| yes | +D (24.40%) | 134 | BC | [143] | poor |
| yes | +D (65.10%) LMaP +D (43.70%) Inv | 43 80 | OC | [145] | limited |
| 60.9% | +D (38.70%) | 266 | CRC | [146] | poor |
| +V (27.44%) +D (58.70%) +B (76.76%) | 164 155 142 | BC | [147] | poor poor | |
| yes | +D | 217 | BC | [148] | relapse |
| Cathepsin | Consensus | Location | Chr | Start | End | Transcript (s) |
|---|---|---|---|---|---|---|
| A | no | Promoer | chr20 | 44519281 | 44519481 | NM_000308,NM_001127695,NM_001167594;NM_080608 |
| no | Promoter | chr20 | 44519541 | 44519741 | NM_000308,NM_001127695,NM_001167594;NM_080608 | |
| no | Promoter | chr20 | 44518781 | 44518981 | NM_000308,NM_001127695,NM_001167594;NM_080608 | |
| B | yes | 1stExonIntron | chr8 | 11711471 | 11711751 | NM_001908 |
| no | 1stExonIntron | chr8 | 11720091 | 11720291 | NM_147781,NM_147783,NM_001908 | |
| no | 1stExonIntron | chr8 | 11724891 | 11725091 | NM_147781,NM_147783,NM_147780,NM_147782,NM_001908 | |
| no | 1stExonIntron | chr8 | 11725391 | 11725591 | NM_147781,NM_147783,NM_147780,NM_147782,NM_001908 | |
| C | no | Intragenic | chr11 | 88060751 | 88060951 | NM_001814,NM_001114173,NM_148170 |
| no | Promoter | chr11 | 88070831 | 88071031 | NM_001114173,NM_001814,NM_148170 | |
| no | Promoter | chr11 | 88070771 | 88071121 | NM_001114173,NM_001814,NM_148170 | |
| D | yes | Intragenic | chr11 | 1778621 | 1778821 | NM_001909 |
| no | 1stExonIntron | chr11 | 1783701 | 1783901 | NM_001909 | |
| no | Intragenic | chr11 | 1779141 | 1779341 | NM_001909 | |
| G | no | 1stExonIntron | chr14 | 25044751 | 25044951 | NM_001911 |
| H | no | Intragenic | chr15 | 79222291 | 79222491 | NM_004390 |
| no | Intragenic | chr15 | 79223191 | 79223541 | NM_004390 | |
| no | Promoter | chr15 | 79237871 | 79238071 | NM_004390 | |
| no | Promoter | chr15 | 79238121 | 79238321 | NM_004390 | |
| no | Promoter | chr15 | 79241991 | 79242191 | NM_004390 | |
| L | no | Promoter | chr9 | 90340861 | 90341061 | NM_001257971,NM_001257972,NM_001257973,NM_001912,NM_145918 |
| no | 1stExonIntron | chr9 | 90341261 | 90341461 | NM_001257971,NM_001257972,NM_001257973,NM_001912,NM_145918 | |
| O | no | 1stExonIntron | chr4 | 156874401 | 156874601 | NM_001334 |
| S | yes | Intragenic | chr1 | 150720361 | 150720591 | NM_001199739,NM_004079 |
| no | 1stExonIntron | chr1 | 150738161 | 150738361 | NM_001199739,NM_004079 |
| Cathepsin | Start | End | Strand | Sequence | Cathepsin | Start | End | Strand | Sequence |
|---|---|---|---|---|---|---|---|---|---|
| B | 1386 | 1410 | + | aggcgggagtacaggCATGtctctg | H | 2520 | 2544 | + | ggagCGAGgtggggacaggcaggga |
| CCDS5986 | 1395 | 1419 | − | gcctgtcttcagagaCATGcctgta | CCDS10308 | 2552 | 2576 | + | ggtgCAAGgtgaagacaggcaggga |
| 2136 | 2160 | + | gaatacaactggggtCATGcctgct | 2562 | 2586 | + | gaagacaggcagggaCATGgtgtga | ||
| C | 353 | 377 | + | ctaaCAAAttagctacaagattaga | 2571 | 2595 | − | gtccccacctcacacCATGtccctg | |
| CCDS8282 | 1967 | 1991 | − | tactaggtgtcaggcCCTGtggaca | 2589 | 2613 | − | cccctcatgtcccttCCTGtcccca | |
| 2280 | 2304 | − | tctggcacgcacacaCATGgcgctc | 2590 | 2614 | + | ggggaCAGGaagggacatgaggggt | ||
| 2281 | 2305 | + | agcgcCATGtgtgtgcgtgccagag | K | 2182 | 2206 | + | gagaagctcatgtgaCTTGtcctag | |
| D | 5 | 29 | + | ggtgcCAGGtccaggctggccgtgg | CCDS969 | 2191 | 2215 | − | atccccaatctaggaCAAGtcacat |
| CCDS7725 | 506 | 530 | − | acaaatcatttaaggCAGGtccaag | L | 92 | 116 | − | caccaggaggggtggCATGttcacc |
| 784 | 808 | + | gtgcCACGttggagacaggcctcca | CCDS6675 | 1353 | 1377 | + | gagttCAAGaccagtctggtcaata | |
| 943 | 967 | + | cacattggagatgggCAAGtctggg | 2172 | 2196 | + | atctCAAGagaacgacttggttacc | ||
| 952 | 976 | − | ctcccttagcccagaCTTGcccatc | O | 2469 | 2493 | − | agccaCCTGgcctgccctgtgagcc | |
| 1378 | 1402 | − | taaaaccaggccgggCATGgtgact | CCDS3794 | |||||
| 1379 | 1403 | + | gtcacCATGcccggcctggttttac | S | 248 | 272 | − | actcaCTTGcccaggctggagtgca | |
| 1663 | 1687 | − | gagatggtgtttcacCATGttggcc | CCDS968 | 604 | 628 | + | ggctacaaacacaaaCATGtctact | |
| F | 266 | 290 | + | tttttgaaacagggtCTTGccctgt | 613 | 637 | − | ctcagctgtagtagaCATGtttgtg | |
| CCDS8144 | 436 | 460 | + | gaaatggggttttgcCATGttgccc | 1132 | 1156 | − | ccacgtatggtaaggCAAGtcatct | |
| 525 | 549 | − | ctgggcatggtggctCATGcctata | 1788 | 1812 | − | accatCTTGgccaggctggtcttga | ||
| 526 | 550 | + | ataggcatgagccacCATGcccagc | 1846 | 1870 | − | acaggcacctgccagCATGtccagc | ||
| 617 | 641 | − | gaagtggaagttaggCATGtttcat | 2402 | 2426 | + | ttaaaagcagtaagaCAGGttttcc | ||
| 1040 | 1064 | − | tgtggcatggcaggtCTTGtgtcag | 2658 | 2666 | + | aCTACaagc | ||
| 1041 | 1065 | + | tgacacaagacctgcCATGccacac | 2660 | 2684 | − | ctgggcatggtggtgCATGcttgta | ||
| 1552 | 1576 | + | aaagttaccttggccCATGcccagg | 2948 | 2972 | + | gagtacctcatgtgaCAAGttccaa | ||
| 1562 | 1586 | + | tggccCATGcccaggaatgagtgaa | V | 662 | 686 | − | ccaggcatggtgatgCATGcctgta | |
| 1809 | 1833 | − | ctgggCATGgtggcacgtgcctgta | CCDS6723 | 663 | 687 | + | acaggcatgcatcacCATGcctggc | |
| 1810 | 1834 | + | acaggcacgtgccacCATGcccagc | 1377 | 1399 | − | gaATAAtatccacagtttttact | ||
| H | 1370 | 1394 | − | caccttgcaaagtggCATGttgttg | W | 246 | 270 | − | acctCAAGcaatccacctgccttgg |
| CCDS10308 | 1824 | 1848 | + | ggtgCAAGgtggagacacgcaggga | CCDS8117 | 773 | 797 | − | tgggccccagccagtCTTGtcctgt |
| 1833 | 1857 | − | cccctcatgtccctgCGTGtctcca | 1486 | 1506 | + | agcccttgACCTcacaagtca | ||
| 1834 | 1858 | + | ggagacacgcagggaCATGaggggg | 1945 | 1969 | − | aaaaCAAAaccaggccaggcacggt | ||
| 1889 | 1913 | + | ggtgCAAGgcggggacaggcaggga | 2301 | 2325 | − | gctaggcaTGAGtcaggctcgctag | ||
| 1993 | 2017 | − | acccccatgtctctgCCTGtcccca | 2671 | 2695 | − | acaggcatgcaccacCATGcccagc | ||
| 1994 | 2018 | + | ggggacaggcagagaCATGggggtg | 2672 | 2696 | + | ctgggcatggtggtgCATGcctgta | ||
| 2116 | 2140 | − | ccccccatgtccctgCCTGtctcca | Z | 102 | 126 | + | cgcccccacaaggaaCATGtttaag | |
| 2117 | 2141 | + | ggagacaggcagggaCATGgggggt | CCDS13474 | 111 | 135 | − | atgcaagatcttaaaCATGttcctt | |
| 2180 | 2204 | − | ctccccatgtccctgCTTGtcccca | 276 | 300 | − | ctagtcgagtggatgCATGtctggc | ||
| 2181 | 2205 | + | ggggacaagcagggaCATGgggagt | 1227 | 1251 | − | tcagCAAGgcaggcacacgacccct | ||
| 2212 | 2236 | − | cgccccacgtccctgCCTGtcccca | 1786 | 1810 | + | caccgccagctcaagCTTGgggact | ||
| 2275 | 2299 | − | ctccccatgtccctgCCTGtctcca | 1872 | 1896 | − | ctcttatttgtttggCAAGtcgctc | ||
| 2276 | 2300 | + | ggagacaggcagggaCATGgggagt | ||||||
| 2404 | 2428 | + | ggggacaggcagggaCATGggggtg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soond, S.M.; Savvateeva, L.V.; Makarov, V.A.; Gorokhovets, N.V.; Townsend, P.A.; Zamyatnin, A.A., Jr. Making Connections: p53 and the Cathepsin Proteases as Co-Regulators of Cancer and Apoptosis. Cancers 2020, 12, 3476. https://doi.org/10.3390/cancers12113476
Soond SM, Savvateeva LV, Makarov VA, Gorokhovets NV, Townsend PA, Zamyatnin AA Jr. Making Connections: p53 and the Cathepsin Proteases as Co-Regulators of Cancer and Apoptosis. Cancers. 2020; 12(11):3476. https://doi.org/10.3390/cancers12113476
Chicago/Turabian StyleSoond, Surinder M., Lyudmila V. Savvateeva, Vladimir A. Makarov, Neonila V. Gorokhovets, Paul A. Townsend, and Andrey A. Zamyatnin, Jr. 2020. "Making Connections: p53 and the Cathepsin Proteases as Co-Regulators of Cancer and Apoptosis" Cancers 12, no. 11: 3476. https://doi.org/10.3390/cancers12113476