Construction of a Novel Multigene Panel Potently Predicting Poor Prognosis in Patients with Clear Cell Renal Cell Carcinoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
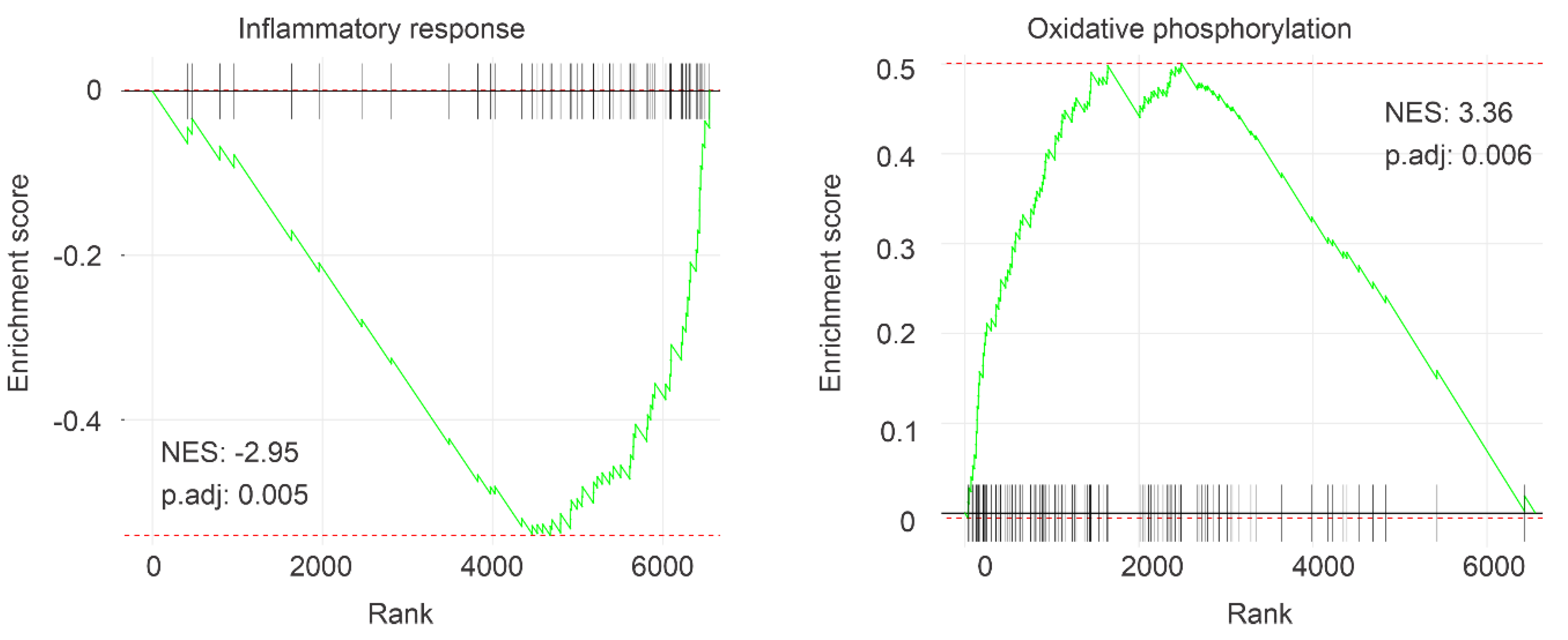
2.1. Association of IQGAP1 Downregulation with Decreases of OS in ccRCC Likely via Impacts on Multiple Processes
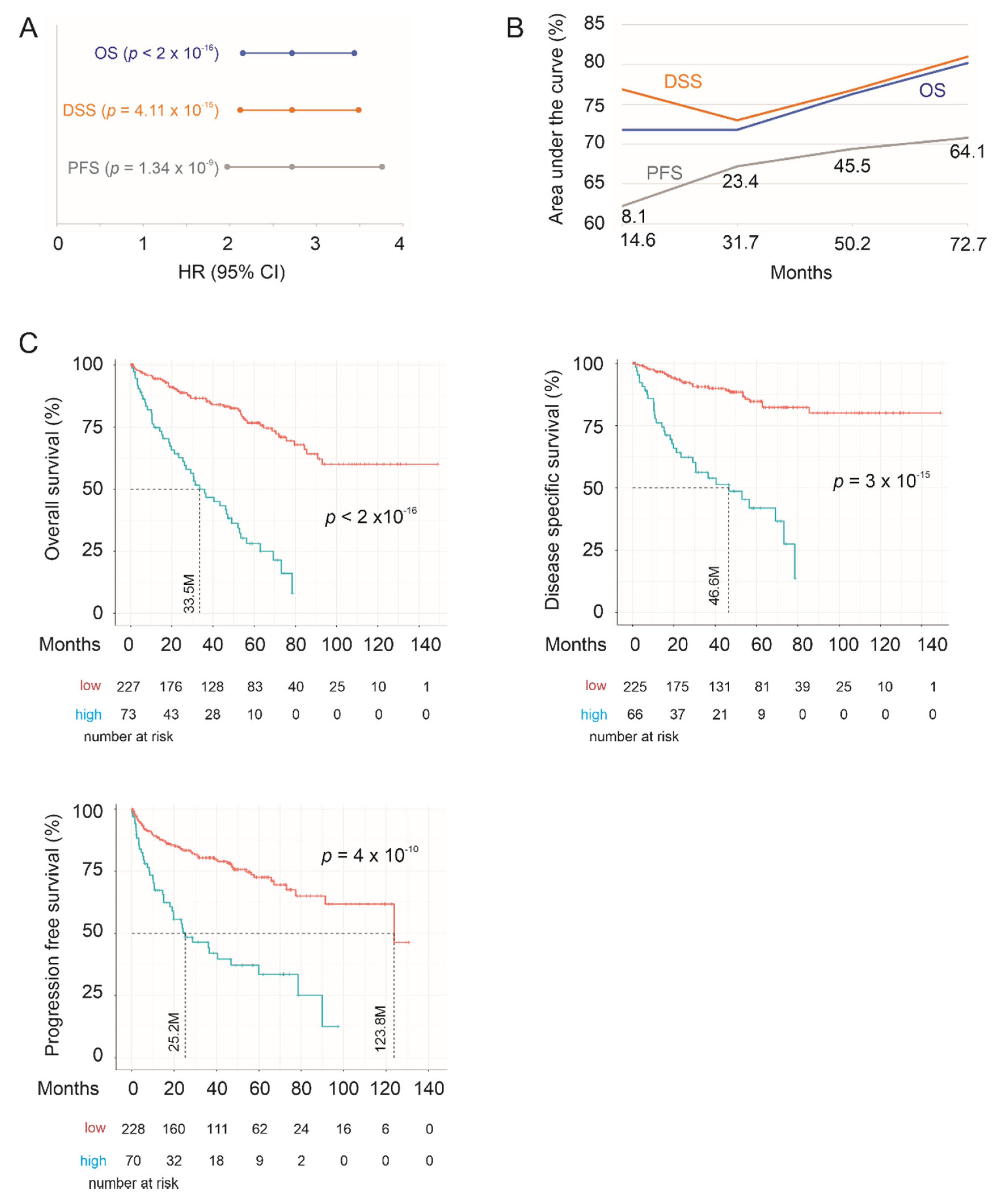
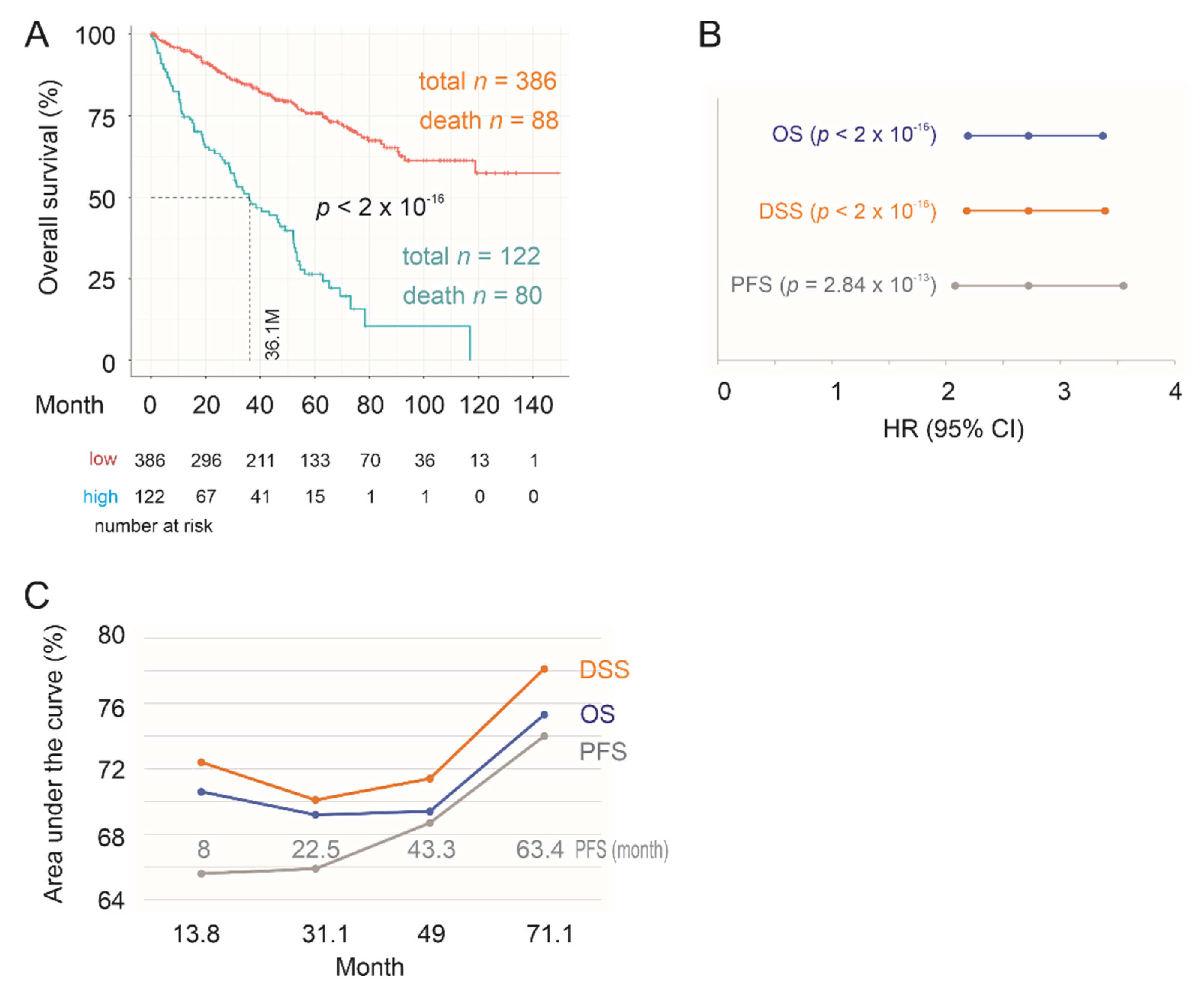
2.2. Construction of SigIQGAP1NW to Predict OS in ccRCC Following Nephrectomy
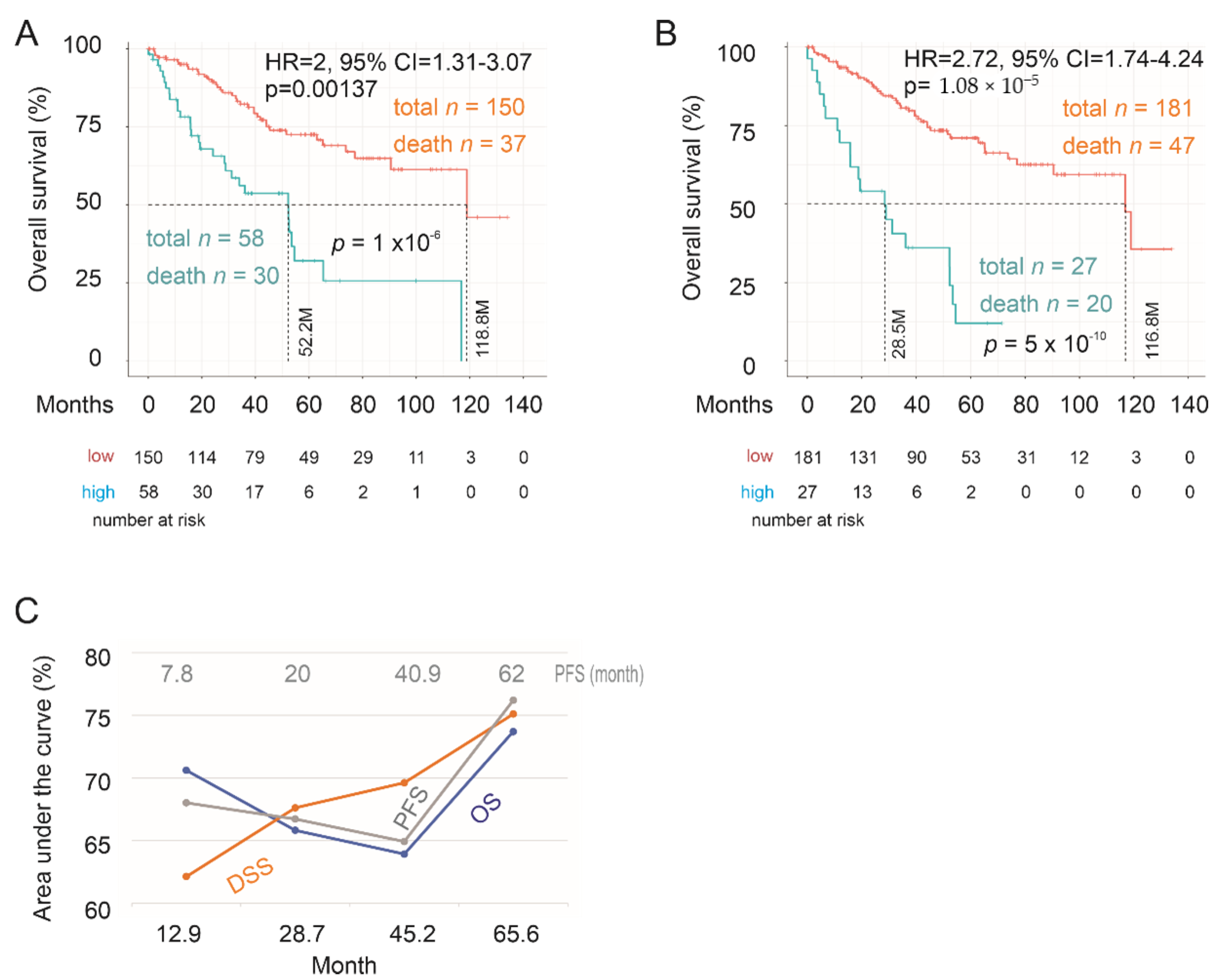
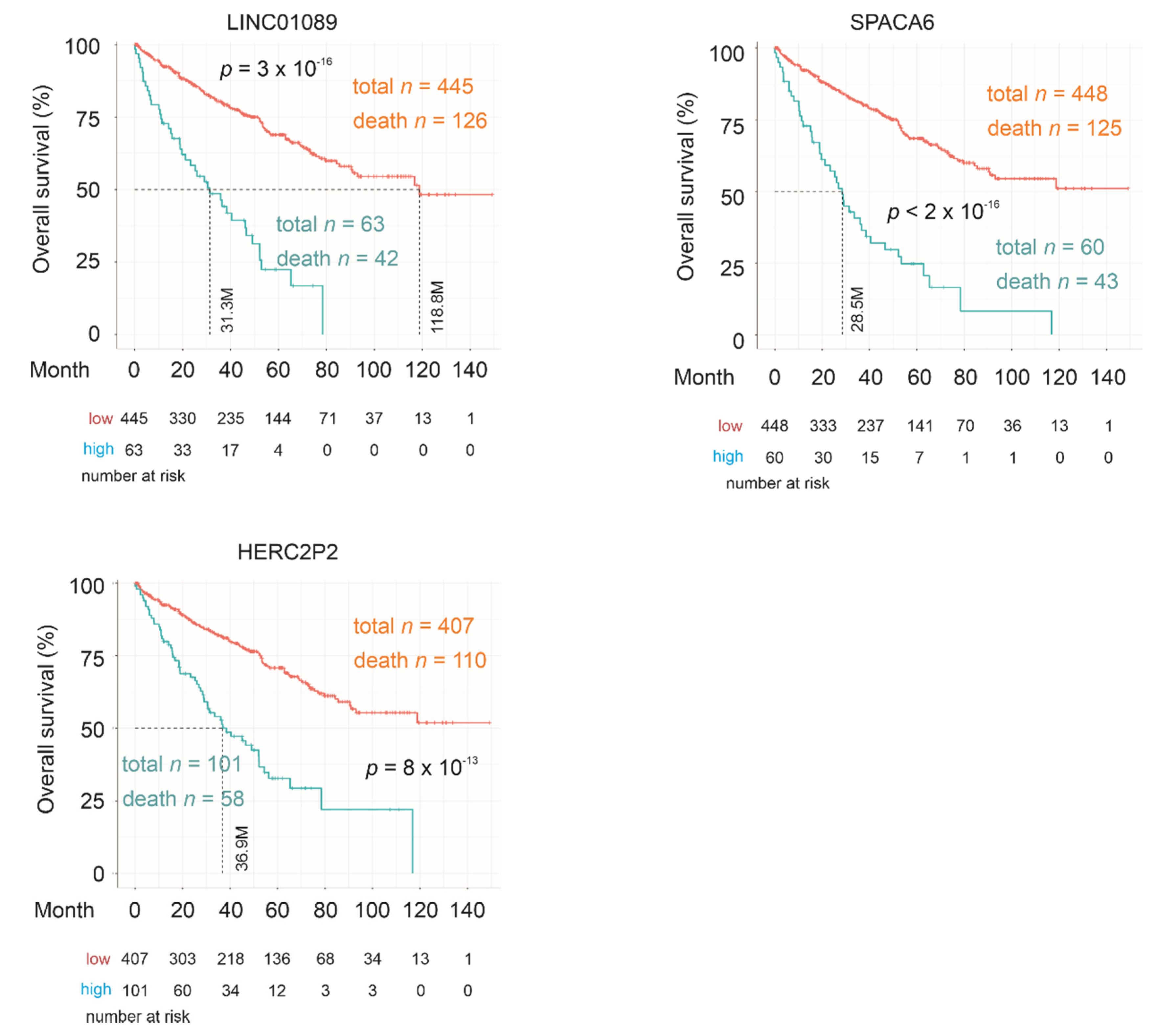
2.3. Testing SigIQGAP1NW
2.4. Association of SigIQGAP1NW with Worse Clinical Features of ccRCC
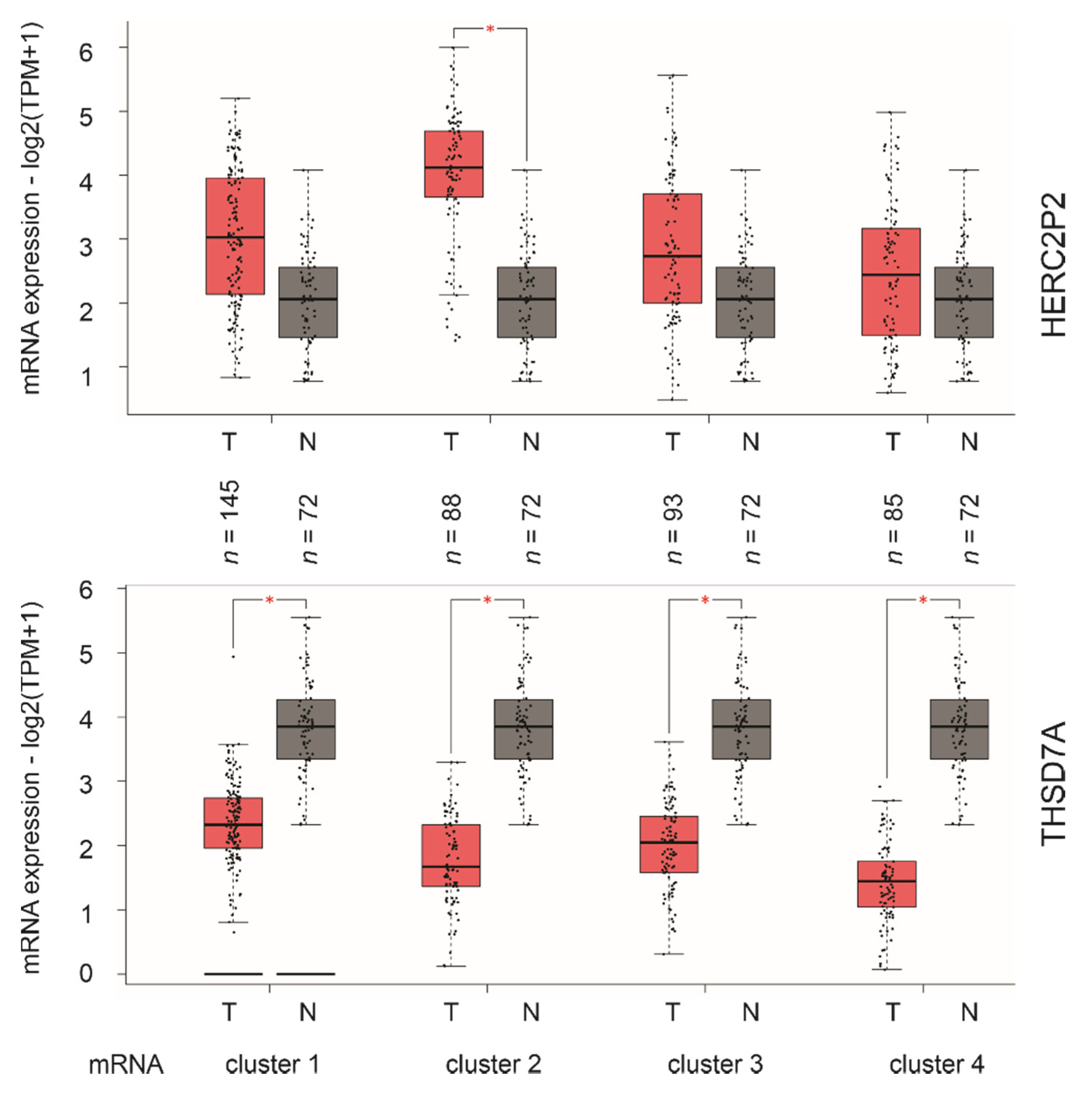
2.5. SigIQGAP1NW, a Novel Multigene Panel of ccRCC Biomarker
3. Discussion
4. Materials and Methods
4.1. Patient Populations
4.2. Pathway Enrichment Analysis
4.3. Regression Analyses
4.4. Construction of Multigene Signatures
4.5. Assignment of Signature Scores to Patients/Tumors
4.6. Cutoff Point Estimation
4.7. Odds Ratio Determination
4.8. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Medina-Rico, M.; Ramos, H.L.; Lobo, M.; Romo, J.; Prada, J.G. Epidemiology of renal cancer in developing countries: Review of the literature. Can. Urol. Assoc. J. Journal de l’Association des Urologues du Canada 2018, 12, E154–E162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majer, W.; Kluzek, K.; Bluyssen, H.; Wesoly, J. Potential approaches and recent advances in biomarker discovery in clear-cell renal cell carcinoma. J. Cancer 2015, 6, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Park, B.; Hwang, E.C.; Hong, S.H.; Jeong, C.W.; Kwak, C.; Byun, S.S.; Chung, J. Retrospective multicenter long-term follow-up analysis of prognostic risk factors for recurrence-free, metastasis-free, cancer-specific, and overall survival after curative nephrectomy in non-metastatic renal cell carcinoma. Front. Oncol. 2019, 9, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, N.; Drake, C.G. Kidney cancer: An overview of current therapeutic approaches. Urol. Clin. N. Am. 2020, 47, 419–431. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Zbar, B.; Brauch, H.; Talmadge, C.; Linehan, M. Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 1987, 327, 721–724. [Google Scholar] [CrossRef]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the vhl tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M.; et al. Silencing of the vhl tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Gulati, S.; Martinez, P.; Joshi, T.; Birkbak, N.J.; Santos, C.R.; Rowan, A.J.; Pickering, L.; Gore, M.; Larkin, J.; Szallasi, Z.; et al. Systematic evaluation of the prognostic impact and intratumour heterogeneity of clear cell renal cell carcinoma biomarkers. Eur. Urol. 2014, 66, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M.; Hedman, A.C.; Sacks, D.B. Iqgaps choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 2015, 25, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Kapoor, A.; Peng, H.; Cutz, J.C.; Tao, L.; Tang, D. Iqgap2 displays tumor suppression functions. J. Anal. Oncol. 2015, 4, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Yan, J.; Cutz, J.C.; Rybak, A.P.; He, L.; Wei, F.; Kapoor, A.; Schmidt, V.A.; Tao, L.; Tang, D. Iqgap2, a candidate tumour suppressor of prostate tumorigenesis. Biochim. Biophys. Acta 2012, 1822, 875–884. [Google Scholar] [CrossRef] [Green Version]
- Hensel, J.; Duex, J.E.; Owens, C.; Dancik, G.M.; Edwards, M.G.; Frierson, H.F.; Theodorescu, D. Patient Mutation Directed shRNA Screen Uncovers Novel Bladder Tumor Growth Suppressors. Mol. Cancer Res. 2015, 13, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Billadeau, D.D.; Abdelhakim, H.; Leof, E.; Kaibuchi, K.; Bernabeu, C.; Bloom, G.S.; Yang, L.; Boardman, L.; Shah, V.H.; et al. Iqgap1 suppresses tbetarii-mediated myofibroblastic activation and metastatic growth in liver. J. Clin. Investig. 2013, 123, 1138–1156. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated tcga pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416 e411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. Gepia2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A cerna hypothesis: The rosetta stone of a hidden rna language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Urrutia, E.; Bustamante Montes, L.P.; Ladron de Guevara Cervantes, D.; Perez-Plasencia, C.; Campos-Parra, A.D. Crosstalk between long non-coding rnas, micro-rnas and mrnas: Deciphering molecular mechanisms of master regulators in cancer. Front. Oncol. 2019, 9, 669. [Google Scholar] [CrossRef]
- Liu, B.; Li, J.; Cairns, M.J. Identifying mirnas, targets and functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Barth, D.A.; Slaby, O.; Klec, C.; Juracek, J.; Drula, R.; Calin, G.A.; Pichler, M. Current concepts of non-coding rnas in the pathogenesis of non-clear cell renal cell carcinoma. Cancers 2019, 11, 1580. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Qin, Y.; Zeng, B.; Feng, Y.; Li, Y.; Xiang, T.; Ren, G. Long noncoding rna linc01089 predicts clinical prognosis and inhibits cell proliferation and invasion through the wnt/beta-catenin signaling pathway in breast cancer. OncoTarg. Ther. 2019, 12, 4883–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas-Chen, A.; Aure, M.R.; Leibovich, L.; Carvalho, S.; Enuka, Y.; Korner, C.; Polycarpou-Schwarz, M.; Lavi, S.; Nevo, N.; Kuznetsov, Y.; et al. Limt is a novel metastasis inhibiting lncrna suppressed by egf and downregulated in aggressive breast cancer. EMBO Mol. Med. 2016, 8, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Yuan, K.; Yan, X.; Xu, L.; Liao, H.; Hao, X.; Wang, J.; Liu, H.; Chen, X.; Xie, K.; et al. Lncrna snhg10 facilitates hepatocarcinogenesis and metastasis by modulating its homolog scarna13 via a positive feedback loop. Cancer Res. 2019, 79, 3220–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.; Lyu, Y.; Jiang, J.; Wang, M.; Zhang, R.; Liew, C.C.; Wang, B.; Cheng, C. Peripheral blood transcriptome identifies high-risk benign and malignant breast lesions. PLoS ONE 2020, 15, e0233713. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. Recq helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef]
- Su, Y.; Meador, J.A.; Calaf, G.M.; Proietti De-Santis, L.; Zhao, Y.; Bohr, V.A.; Balajee, A.S. Human recql4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010, 70, 9207–9217. [Google Scholar] [CrossRef] [Green Version]
- Mo, D.; Fang, H.; Niu, K.; Liu, J.; Wu, M.; Li, S.; Zhu, T.; Aleskandarany, M.A.; Arora, A.; Lobo, D.N.; et al. Human helicase recql4 drives cisplatin resistance in gastric cancer by activating an akt-yb1-mdr1 signaling pathway. Cancer Res. 2016, 76, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The biology of iqgap proteins: Beyond the cytoskeleton. EMBO Rep. 2015, 16, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Hu, S.L.; Chang, A.; Perazella, M.A.; Okusa, M.D.; Jaimes, E.A.; Weiss, R.H.; American Society of Nephrology Onco-Nephrology, F. The nephrologist’s tumor: Basic biology and management of renal cell carcinoma. J. Am. Soc. Nephrol. JASN 2016, 27, 2227–2237. [Google Scholar] [CrossRef] [Green Version]
- Schepisi, G.; Farolfi, A.; Conteduca, V.; Martignano, F.; De Lisi, D.; Ravaglia, G.; Rossi, L.; Menna, C.; Bellia, S.R.; Barone, D.; et al. Immunotherapy for prostate cancer: Where we are headed. Int. J. Mol. Sci. 2017, 18, 2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farolfi, A.; Gurioli, G.; Fugazzola, P.; Burgio, S.L.; Casanova, C.; Ravaglia, G.; Altavilla, A.; Costantini, M.; Amadori, A.; Framarini, M.; et al. Immune system and DNA repair defects in ovarian cancer: Implications for locoregional approaches. Int. J. Mol. Sci. 2019, 20, 2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.D.; Brown, M.D.; Sacks, D.B. Iqgaps in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeCour, L., Jr.; Boyapati, V.K.; Liu, J.; Li, Z.; Sacks, D.B.; Worthylake, D.K. The structural basis for cdc42-induced dimerization of iqgaps. Structure 2016, 24, 1499–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jameson, K.L.; Mazur, P.K.; Zehnder, A.M.; Zhang, J.; Zarnegar, B.; Sage, J.; Khavari, P.A. Iqgap1 scaffold-kinase interaction blockade selectively targets ras-map kinase-driven tumors. Nat. Med. 2013, 19, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Aakula, A.; Kohonen, P.; Leivonen, S.K.; Makela, R.; Hintsanen, P.; Mpindi, J.P.; Martens-Uzunova, E.; Aittokallio, T.; Jenster, G.; Perala, M.; et al. Systematic identification of micrornas that impact on proliferation of prostate cancer cells and display changed expression in tumor tissue. Eur. Urol. 2016, 69, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Xu, H.; Major, P.; Tang, D. Assessment of biochemical recurrence of prostate cancer (Review). Int. J. Oncol. 2019, 55, 1194–1212. [Google Scholar] [CrossRef] [Green Version]
- Inoue, N.; Hagihara, Y.; Wright, D.; Suzuki, T.; Wada, I. Oocyte-triggered dimerization of sperm izumo1 promotes sperm-egg fusion in mice. Nat. Commun. 2015, 6, 8858. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Ye, Y.; Aakre, J.A.; Pu, X.; Chang, G.C.; Yang, P.C.; Roth, J.A.; Marks, R.S.; Lippman, S.M.; et al. Genome-wide association study of genetic predictors of overall survival for non-small cell lung cancer in never smokers. Cancer Res. 2013, 73, 4028–4038. [Google Scholar] [CrossRef] [Green Version]
- Colombo, E.A.; Locatelli, A.; Cubells Sanchez, L.; Romeo, S.; Elcioglu, N.H.; Maystadt, I.; Esteve Martinez, A.; Sironi, A.; Fontana, L.; Finelli, P.; et al. Rothmund-thomson syndrome: Insights from new patients on the genetic variability underpinning clinical presentation and cancer outcome. Int. J. Mol. Sci. 2018, 19, 1103. [Google Scholar] [CrossRef] [Green Version]
- Carril-Ajuria, L.; Santos, M.; Roldan-Romero, J.M.; Rodriguez-Antona, C.; de Velasco, G. Prognostic and predictive value of pbrm1 in clear cell renal cell carcinoma. Cancers 2019, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas, N.M.; Beck, L.H., Jr.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.S.; et al. Thrombospondin type-1 domain-containing 7a in idiopathic membranous nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, A.F.; Jegede, O.; Haas, N.B.; Flaherty, K.T.; Pins, M.R.; Messing, E.M.; Manola, J.; Wood, C.G.; Kane, C.J.; Jewett, M.A.S.; et al. Predicting renal cancer recurrence: Defining limitations of existing prognostic models with prospective trial-based validation. J. Clin. Oncol. 2019, 37, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Kattan, M.W.; Reuter, V.; Motzer, R.J.; Katz, J.; Russo, P. A postoperative prognostic nomogram for renal cell carcinoma. J. Urol. 2001, 166, 63–67. [Google Scholar] [CrossRef]
- Yaycioglu, O.; Roberts, W.W.; Chan, T.; Epstein, J.I.; Marshall, F.F.; Kavoussi, L.R. Prognostic assessment of nonmetastatic renal cell carcinoma: A clinically based model. Urology 2001, 58, 141–145. [Google Scholar] [CrossRef]
- Zisman, A.; Pantuck, A.J.; Wieder, J.; Chao, D.H.; Dorey, F.; Said, J.W.; deKernion, J.B.; Figlin, R.A.; Belldegrun, A.S. Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J. Clin. Oncol. 2002, 20, 4559–4566. [Google Scholar] [CrossRef]
- Frank, I.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Weaver, A.L.; Zincke, H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: The ssign score. J. Urol. 2002, 168, 2395–2400. [Google Scholar] [CrossRef]
- Cindolo, L.; de la Taille, A.; Messina, G.; Romis, L.; Abbou, C.C.; Altieri, V.; Rodriguez, A.; Patard, J.J. A preoperative clinical prognostic model for non-metastatic renal cell carcinoma. BJU Int. 2003, 92, 901–905. [Google Scholar] [CrossRef]
- Leibovich, B.C.; Blute, M.L.; Cheville, J.C.; Lohse, C.M.; Frank, I.; Kwon, E.D.; Weaver, A.L.; Parker, A.S.; Zincke, H. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: A stratification tool for prospective clinical trials. Cancer 2003, 97, 1663–1671. [Google Scholar] [CrossRef]
- Sorbellini, M.; Kattan, M.W.; Snyder, M.E.; Reuter, V.; Motzer, R.; Goetzl, M.; McKiernan, J.; Russo, P. A postoperative prognostic nomogram predicting recurrence for patients with conventional clear cell renal cell carcinoma. J. Urol. 2005, 173, 48–51. [Google Scholar] [CrossRef] [Green Version]
- Karakiewicz, P.I.; Briganti, A.; Chun, F.K.; Trinh, Q.D.; Perrotte, P.; Ficarra, V.; Cindolo, L.; De la Taille, A.; Tostain, J.; Mulders, P.F.; et al. Multi-institutional validation of a new renal cancer-specific survival nomogram. J. Clin. Oncol. 2007, 25, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pan, Q.; Xiao, R.; Yu, Y.; Lu, W.; Wang, L. A cluster of metabolism-related genes predict prognosis and progression of clear cell renal cell carcinoma. Sci. Rep. 2020, 10, 12949. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Dang, W.; Wang, S.; Hu, B.; Yin, L.; Guan, B. An optimal prognostic model based on gene expression for clear cell renal cell carcinoma. Oncol. Lett. 2020, 20, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiang, Z.; Chen, X.; Zhu, X.; Peng, X. A seven-gene signature model predicts overall survival in kidney renal clear cell carcinoma. Hereditas 2020, 157, 38. [Google Scholar] [CrossRef]
- Verbiest, A.; Van Hoef, V.; Rodriguez-Antona, C.; Garcia-Donas, J.; Grana-Castro, O.; Albersen, M.; Baldewijns, M.; Laenen, A.; Roussel, E.; Schoffski, P.; et al. Microrna expression profiles in molecular subtypes of clear-cell renal cell carcinoma are associated with clinical outcome and repression of specific mrna targets. PLoS ONE 2020, 15, e0238809. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | P.Adj | ES | NES | Size |
|---|---|---|---|---|
| INFγ response | 0.005545 | −0.37972 | −2.2396 | 79 |
| Il2_Stat5 signaling | 0.005545 | −0.39412 | −2.3246 | 79 |
| Mitotic spindle | 0.005545 | −0.36571 | −2.3260 | 115 |
| UV response DN (down) | 0.005545 | −0.4882 | −2.8751 | 78 |
| Allograft rejection | 0.005545 | −0.4118 | −2.3087 | 67 |
| TNFα signaling via NFκB | 0.005545 | −0.48317 | −2.5962 | 57 |
| Estrogen response early | 0.005545 | −0.39141 | −2.1554 | 63 |
| Complement | 0.005545 | −0.36543 | −2.1273 | 76 |
| Apical junction | 0.005545 | −0.41499 | −2.3820 | 73 |
| Epithelial mesenchymal transition | 0.005545 | −0.56703 | −3.0 | 62 |
| KRAS signaling UP | 0.005545 | −0.60277 | −3.2622 | 59 |
| Inflammatory response | 0.005545 | −0.54082 | −2.9457 | 61 |
| TGFβ signaling | 0.005545 | −0.50114 | −2.1570 | 26 |
| Androgen response | 0.005545 | −0.44591 | −2.2861 | 50 |
| Coagulation | 0.005545 | −0.46409 | −2.1825 | 37 |
| Oxidative phosphorylation | 0.006152 | 0.50062 | 3.3574 | 118 |
| DNA repair | 0.011239 | 0.29602 | 1.8111 | 83 |
| Adipogenesis | 0.011239 | 0.28891 | 1.8073 | 91 |
| IL6 Jak Stat3 signaling | 0.012913 | −0.43411 | −1.9242 | 29 |
| Fatty acid metabolism | 0.028448 | 0.31956 | 1.7648 | 52 |
| G2M checkpoin | 0.033794 | −0.27572 | −1.6313 | 81 |
| Spermatogenesis | 0.033962 | −0.38359 | −1.7162 | 31 |
| Protein secretion | 0.048784 | −0.28187 | −1.5703 | 66 |
| Gene | Locus | Log2 Ratio 1 | p-Value | q-Value |
|---|---|---|---|---|
| LINC01089 | 12q24.31 | 1.19 | 4.84 × 10−17 | 3.69 × 10−16 |
| SPACA6 | 19q13.41 | 1.12 | 2 × 10−16 | 1.41 × 10−15 |
| LOC155060 | 7q36.1 | 1.09 | 5.01 × 10−11 | 1.90 × 10−10 |
| LOC100128288 | 17p13.1 | 1.08 | 3.54 × 10−19 | 3.72 × 10−18 |
| SNHG10 | 14q32.13 | 1.05 | 8.40 × 10−17 | 6.19 × 10−16 |
| RECQL4 | 8q24.3 | 1.01 | 2.67 × 10−17 | 2.11 × 10−16 |
| HERC2P2 | 15q11.2 | 0.97 | 2.52 × 10−8 | 7.14 × 10−8 |
| ATXN7L2 | 1p13.3 | 0.95 | 3.15 × 10−14 | 1.69 × 10−13 |
| THSD7A | 7p21.3 | −1.44 | 3.38 × 10−14 | 1.81 × 10−13 |
| Factors | Univariate Cox Analysis | Multivariate Cox Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Sig 1 | 2.72 | 2.19–3.37 | <2 × 10−16 *** | 1.80 | 1.41–2.31 | 2.85 × 10−6 *** |
| Age 2 | 1.03 | 1.02–1.04 | 2.78 × 10−6 *** | 1.03 | 1.02–1.05 | 6.34 × 10−5 *** |
| Sex 3 | 0.96 | 0.70–1.31 | 0.793 | 1.08 | 0.77–1.51 | 0.64719 |
| State III 4 | 2.8 | 1.84–4.24 | 1.28 × 10−6 *** | 1.82 | 1.18–2.85 | 0.00714 ** |
| State IV 4 | 6.83 | 4.60–10.12 | <2 × 10−16 *** | 4.49 | 2.85–7.07 | 9.30 × 10−11 *** |
| Grade 3 5 | 1.94 | 1.32–2.86 | 0.00075 *** | 1.22 | 0.81–1.84 | 0.34185 |
| Grade 4 5 | 5.38 | 3.59–8.05 | 3.06 × 10−16 *** | 1.73 | 1.07–2.81 | 0.02540 * |
| WHS 6 | 1.04 | 1.03–1.05 | 2.19 × 10−11 *** | 1.01 | 1.002–1.03 | 0.02525 * |
| Gene | HR | 95% CI | p Value |
|---|---|---|---|
| LINC01089 | 1.002 | 1.001–1.002 | 3.14 × 10−13 *** |
| SPACA6 | 1.006 | 1.005–1.008 | 2.69 × 10−15 *** |
| LOC155060 | 1.002 | 1.001–1.003 | 1.31 × 10−13 *** |
| LOC100128288 | 1.008 | 1.005–1.01 | 4.53 × 10−8 *** |
| SNHG10 | 1.007 | 1.005–1.009 | 5.9 × 10−10 *** |
| RECQL4 | 1.003 | 1.002–1.004 | 9.7 × 10−14 *** |
| HERC2P2 | 1.0 | 1.0–1.001 | 5.64 × 10−11 *** |
| ATXN7L2 | 1.005 | 1.004–1.006 | 5.24 × 10−14 *** |
| THSD7A | 0.9991 | 0.9984–0.9999 | 0.0292 * |
| Gene | Gene Details | ccRCC 1 | Oncogenesis 2 | Refs |
|---|---|---|---|---|
| LINC01089 | long intergenic non-protein-coding RNA 1089 | unknown | inhibition of breast cancer metastasis | [32] |
| SPACA6 | sperm acrosome associated 6 | unknown | unknown | NA |
| LOC155060 | AI894139 pseudogene | unknown | unknown | NA |
| LOC100128288 | uncharacterized, an RNA gene that is affiliated with the lncRNA class | unknown | unknown | NA |
| SNHG10 | small nucleolar RNA host gene 10, a non-protein-coding RNA | unknown | promotion of hepatocellular carcinoma metastasis via activating c-Myb | [33] |
| RECQL4 | RecQ like helicase 4 | unknown | Driving gastric cancer resistance to cisplatin via activating the AKT-YB1-MDR pathway | [37] |
| HERC2P2 | hect domain and RLD 2 pseudogene 2 | unknown | A component of a 10-gene blood biomarker of breast cancer | [34] |
| ATXN7L2 | ataxin 7 like 2 | unknown | Not clear | NA |
| THSD7A | thrombospondin type 1 domain containing 7A | unknown | Not clear | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Major, P.; Tang, D. Construction of a Novel Multigene Panel Potently Predicting Poor Prognosis in Patients with Clear Cell Renal Cell Carcinoma. Cancers 2020, 12, 3471. https://doi.org/10.3390/cancers12113471
Lin X, Kapoor A, Gu Y, Chow MJ, Peng J, Major P, Tang D. Construction of a Novel Multigene Panel Potently Predicting Poor Prognosis in Patients with Clear Cell Renal Cell Carcinoma. Cancers. 2020; 12(11):3471. https://doi.org/10.3390/cancers12113471
Chicago/Turabian StyleLin, Xiaozeng, Anil Kapoor, Yan Gu, Mathilda Jing Chow, Jingyi Peng, Pierre Major, and Damu Tang. 2020. "Construction of a Novel Multigene Panel Potently Predicting Poor Prognosis in Patients with Clear Cell Renal Cell Carcinoma" Cancers 12, no. 11: 3471. https://doi.org/10.3390/cancers12113471