The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir



Abstract
:Simple Summary
Abstract

1. Introduction
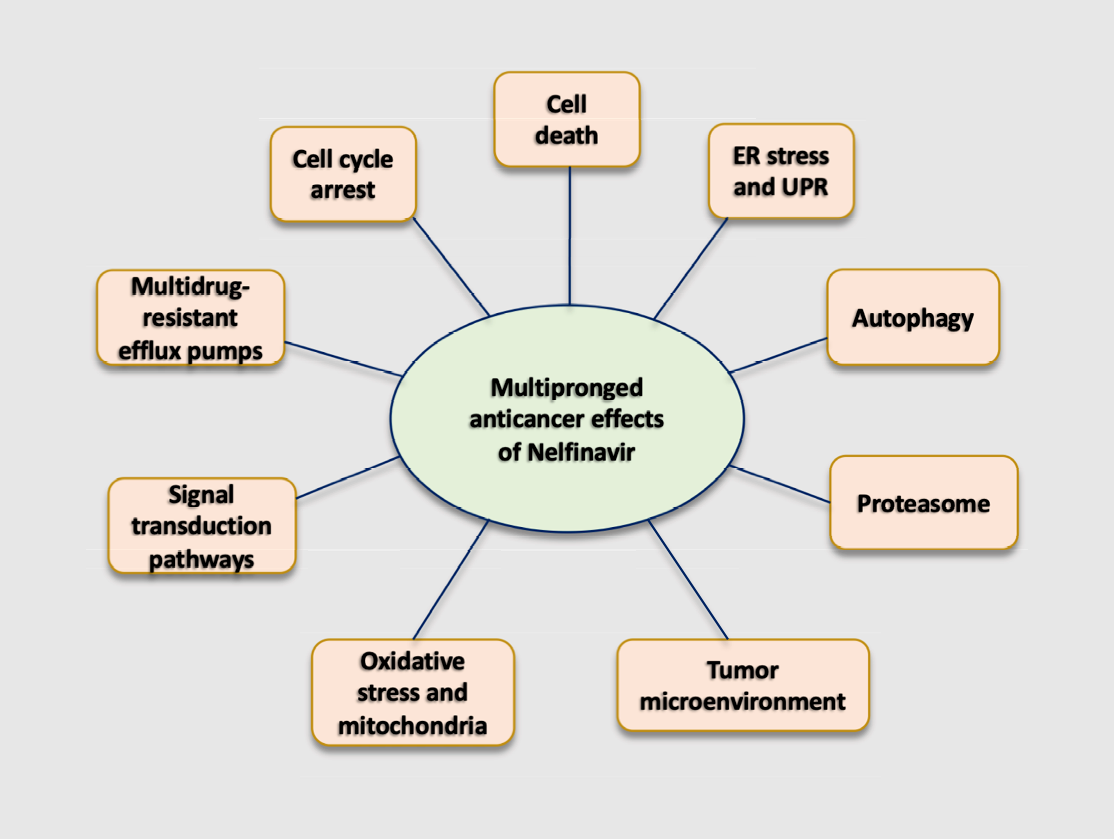
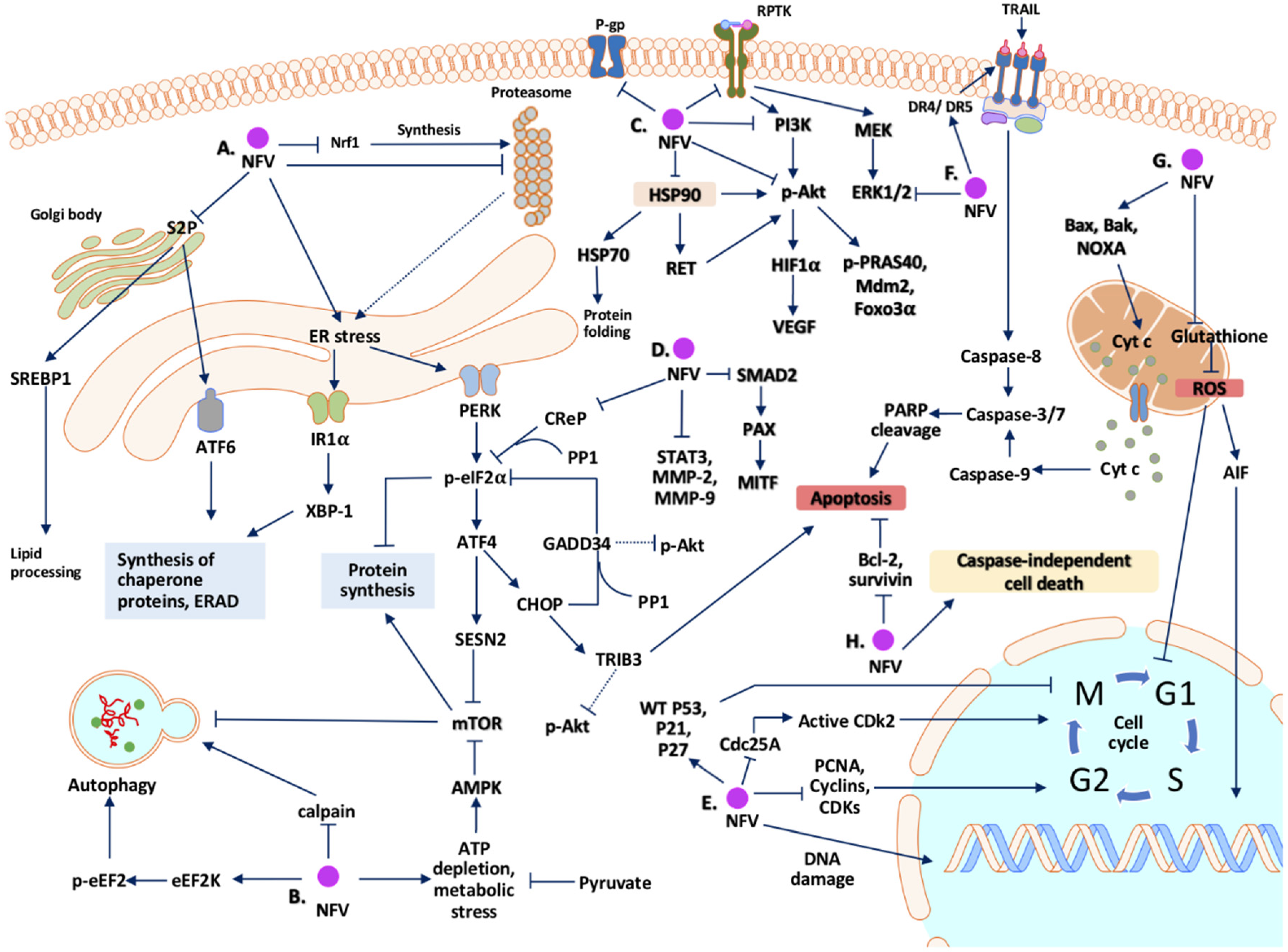
2. Potential Mechanisms Whereby Nelfinavir Exert Its Anti-Cancer Effect
2.1. Cell Cycle Arrest
2.2. Cell Death
2.3. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR)
2.4. Autophagy
2.5. Inhibition of the Proteasome
2.6. Signal Transduction Pathways
2.7. Oxidative Stress and Mitochondria
2.8. Tumor Microenvironment
2.9. Multidrug-Resistant Efflux Pumps
2.10. Summary of Mechanisms of Action of Nelfinavir as an Anti-Cancer Agent
3. Anti-Tumor Effects of Nelfinavir: Preclinical Evidences In Vivo
4. Current Status of Clinical Trials
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AIDS | Acquired immunodeficiency syndrome |
| AIF | Apoptosis-inducing factor |
| AML | Acute myeloid leukemia |
| AMPK | 5′-AMP-activated protein kinase |
| ATF3 | Activating transcription factor 3 |
| ATF6 | Activating transcription factor 6 |
| ATP | Adenosine triphosphate |
| BrdU | Bromodeoxyuridine |
| CDK | Cyclin-dependent kinase |
| CHOP | CCAAT enhancer-binding protein homologous protein |
| CML | Chronic myeloid leukemia |
| CT | Computer tomography |
| DR | Death receptors |
| DEN | Diethylnitrosamine |
| DMC | Dimethylcelecoxib |
| eIF2α | Eukaryotic initiation factor 2α |
| eEF2 | Eukaryotic elongation factor |
| eEF2K | Eukariotic elongation factor 2 kinase |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERp44 | Endoplasmic reticulum resident protein 44 |
| ERO1-Lα | Endoplasmic reticulum oxidoreductin-1-like protein α |
| FACS | Fluorescence-activated cell sorting |
| FADD | Fas-associated protein with death domain |
| FAK | Focal adhesion kinase |
| FAS | Fatty acid synthase |
| FDA | Food and Drug Administration |
| FMISO-PET | 18F-fluoromisonidazole positron emission tomography |
| GADD34 | Growth arrest and DNA damage inducible protein 34 |
| GBM | Glioblastoma multiforme |
| GFP | Green fluorescent protein |
| GRP78 | Glucose-regulated protein of 78 kDa |
| GSH | Glutathione |
| HAART | Highly active antiretroviral treatment |
| HCC | Hepatocarcinoma cells |
| HDAC | Histone deacetylase |
| HER2 | Human epidermal factor receptor 2 |
| HIF1α | Hypoxia-inducible factor 1α |
| HIV | Human immunodeficiency virus |
| HPV | Human papillomavirus |
| hPSC | Human pancreatic stellate cells |
| HSP90 | Heat shock protein 90 |
| H2O2 | Hydrogen peroxide |
| IGFR | Insulin-like growth factor receptor |
| IRE1α | Inositol-requiring enzyme 1-α |
| ISR | Integrated stress response |
| KS | Kaposi’s sarcoma |
| LAPC | Locally advanced pancreatic cancer |
| MAPK | Mitogen-activated protein kinase pathway |
| 3MA | 3-methyladenosine |
| mTOR | Mammalian target of rapamycin |
| 3-MA | 3-methyladenine |
| MDR | Multidrug resistance |
| MDR1 | Multidrug-resistant 1 |
| MRP-4 | Multidrug resistance protein 4 |
| MEF | Mouse embryonic fibroblasts |
| MM | Multiple myeloma |
| mPTP | Mitochondria permeability transition pore |
| MMP-2 | Matrix metalloproteinase-2 |
| MMP-9 | Matrix metalloproteinase-3 |
| NAC | N-acetylcysteine |
| NET | Neuroendocrine tumor |
| NO | Nitric oxide |
| NSCLC | Non-small-cell lung carcinoma |
| PARP | Poly ADP-ribose polymerase |
| PBMC | Peripheral blood mononuclear cells |
| PCNA | Proliferating cell nuclear antigen |
| PDI | Protein disulfide isomerase |
| PE | Phosphatidyl ethanolamine |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| PET | Positron emission tomography |
| P-gp | P-glycoprotein |
| PI | Protease inhibitors |
| PTEN | Phosphate and tensin homologue |
| RIP | Regulated intramembrane proteolysis |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SBRT | Stereotactic body radiotherapy |
| SIRT3 | Sirtuin-3 |
| STAT3 | Signal transducer and activator of transcription 3 |
| S1P | Site-1 protease |
| S2P | Site-2 protease |
| SESN2 | Sestrin-2 |
| SRC | Tyrosine kinase Src |
| SREBP1 | Sterol regulatory binding protein-1 |
| siRNA | Small interference RNA |
| TNBC | Triple-negative breast cancer |
| TNF | Tumor necrosis factor |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| TRIB-3 | Tribbles homolog-3 |
| TSC | Tuberous sclerosis complex |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling |
| UPR | Unfolded protein response |
| VEGF | Vascular endothelial growth factor |
| XBP-1 | X-box binding protein-1 |
References
- Flexner, C. HIV-protease inhibitors. N. Engl. J. Med. 1998, 338, 1281–1292. [Google Scholar] [CrossRef]
- Debouck, C. The HIV-1 protease as a therapeutic target for AIDS. AIDS Res. Hum. Retrovir. 1992, 8, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl) 2015, 7, 95–104. [Google Scholar] [PubMed] [Green Version]
- Maksimovic-Ivanic, D.; Fagone, P.; McCubrey, J.; Bendtzen, K.; Mijatovic, S.; Nicoletti, F. HIV-protease inhibitors for the treatment of cancer: Repositioning HIV protease inhibitors while developing more potent NO-hybridized derivatives? Int. J. Cancer 2017, 140, 1713–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, C.C.; Fischl, M.A.; Hammer, S.M.; Hirsch, M.S.; Jacobsen, D.M.; Katzenstein, D.A.; Montaner, J.S.; Richman, D.D.; Saag, M.S.; Schooley, R.T.; et al. Antiretroviral therapy for HIV infection in 1998: Updated recommendations of the International AIDS Society-USA Panel. JAMA 1998, 280, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.E.; Wu, E.; Patick, A.K.; Kerr, B.; Zorbas, M.; Lankford, A.; Kobayashi, T.; Maeda, Y.; Shetty, B.; Webber, S. Circulating metabolites of the human immunodeficiency virus protease inhibitor nelfinavir in humans: Structural identification, levels in plasma, and antiviral activities. Antimicrob. Agents Chemother. 2001, 45, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Niehues, T.; Horneff, G.; Megahed, M.; Schroten, H.; Wahn, V. Complete regression of AIDS-related Kaposi’s sarcoma in a child treated with highly active antiretroviral therapy. AIDS 1999, 13, 1148–1149. [Google Scholar] [CrossRef]
- Lebbe, C.; Blum, L.; Pellet, C.; Blanchard, G.; Verola, O.; Morel, P.; Danne, O.; Calvo, F. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi’s sarcoma. AIDS 1998, 12, F45–F49. [Google Scholar] [CrossRef]
- Krischer, J.; Rutschmann, O.; Hirschel, B.; Vollenweider-Roten, S.; Saurat, J.H.; Pechere, M. Regression of Kaposi’s sarcoma during therapy with HIV-1 protease inhibitors: A prospective pilot study. J. Am. Acad. Dermatol. 1998, 38, 594–598. [Google Scholar] [CrossRef]
- Sgadari, C.; Barillari, G.; Toschi, E.; Carlei, D.; Bacigalupo, I.; Baccarini, S.; Palladino, C.; Leone, P.; Bugarini, R.; Malavasi, L.; et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat. Med. 2002, 8, 225–232. [Google Scholar] [CrossRef]
- Sgadari, C.; Monini, P.; Barillari, G.; Ensoli, B. Use of HIV protease inhibitors to block Kaposi’s sarcoma and tumour growth. Lancet Oncol. 2003, 4, 537–547. [Google Scholar] [CrossRef]
- Schmidtke, G.; Holzhutter, H.G.; Bogyo, M.; Kairies, N.; Groll, M.; de Giuli, R.; Emch, S.; Groettrup, M. How an inhibitor of the HIV-I protease modulates proteasome activity. J. Biol. Chem. 1999, 274, 35734–35740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, V.B.; Nahata, M.C. Nelfinavir mesylate: A protease inhibitor. Ann. Pharm. 1999, 33, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Koltai, T. Nelfinavir and other protease inhibitors in cancer: Mechanisms involved in anticancer activity. F1000Res 2015, 4, 9. [Google Scholar] [CrossRef]
- Gantt, S.; Casper, C.; Ambinder, R.F. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr. Opin. Oncol. 2013, 25, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, R.; Salahub, D.R. Nelfinavir: A magic bullet to annihilate cancer cells? Cancer Biol. Ther. 2009, 8, 233–235. [Google Scholar] [CrossRef]
- Xie, L.; Evangelidis, T.; Xie, L.; Bourne, P.E. Drug Discovery Using Chemical Systems Biology: Weak Inhibition of Multiple Kinases May Contribute to the Anti-Cancer Effect of Nelfinavir. PLoS Comput. Biol. 2011, 7, e1002037. [Google Scholar] [CrossRef]
- Arodola, O.A.; Soliman, M.E. Could the FDA-approved anti-HIV PR inhibitors be promising anticancer agents? An answer from enhanced docking approach and molecular dynamics analyses. Drug Des. Devel. Ther. 2015, 9, 6055–6065. [Google Scholar]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.S.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Driessen, C.; Muller, R.; Novak, U.; Cantoni, N.; Betticher, D.; Mach, N.; Rufer, A.; Mey, U.; Samaras, P.; Ribi, K.; et al. Promising activity of nelfinavir-bortezomib-dexamethasone in proteasome inhibitor-refractory multiple myeloma. Blood 2018, 132, 2097–2100. [Google Scholar] [CrossRef]
- Bruning, A.; Burger, P.; Vogel, M.; Rahmeh, M.; Gingelmaiers, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol. Ther. 2009, 8, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Rahmeh, M.; Gingelmaier, A.; Friese, K. The mitochondria-independent cytotoxic effect of nelfinavir on leukemia cells can be enhanced by sorafenib-mediated mcl-1 downregulation and mitochondrial membrane destabilization. Mol. Cancer 2010, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Vogel, M.; Mylonas, I.; Friese, K.; Burges, A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr. Cancer Drug Targets 2011, 11, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Chow, W.A.; Guo, S.; Valdes-Albini, F. Nelfinavir induces liposarcoma apoptosis and cell cycle arrest by upregulating sterol regulatory element binding protein-1. Anticancer Drugs 2006, 17, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Mikochik, P.J.; Ra, J.H.; Lei, H.; Flaherty, K.T.; Winkler, J.D.; Spitz, F.R. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Res. 2007, 67, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.; Bikas, A.; Patel, A.; Kushchayeva, Y.; Costello, J.; McDaniel, D.; Burman, K.; Vasko, V. Nelfinavir inhibits proliferation and induces DNA damage in thyroid cancer cells. Endocr. Relat. Cancer 2017, 24, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Asano, T.; Okubo, K.; Isono, M.; Asano, T. Nelfinavir and Ritonavir Kill Bladder Cancer Cells Synergistically by Inducing Endoplasmic Reticulum Stress. Oncol. Res. 2018, 26, 323–332. [Google Scholar] [CrossRef]
- Okubo, K.; Sato, A.; Isono, M.; Asano, T.; Asano, T. Nelfinavir Induces Endoplasmic Reticulum Stress and Sensitizes Renal Cancer Cells to TRAIL. Anticancer Res. 2018, 38, 4505–4514. [Google Scholar] [CrossRef]
- Okubo, K.; Isono, M.; Asano, T.; Sato, A. Panobinostat and Nelfinavir Inhibit Renal Cancer Growth by Inducing Endoplasmic Reticulum Stress. Anticancer Res. 2018, 38, 5615–5626. [Google Scholar] [CrossRef]
- Soprano, M.; Sorriento, D.; Rusciano, M.R.; Maione, A.S.; Limite, G.; Forestieri, P.; D’Angelo, D.; D’Alessio, M.; Campiglia, P.; Formisano, P.; et al. Oxidative Stress Mediates the Antiproliferative Effects of Nelfinavir in Breast Cancer Cells. PLoS ONE 2016, 11, e0155970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Niu, L.; Zhu, X.; Hao, J.; Wang, P.; Wang, H. Antitumour effects of a protease inhibitor, nelfinavir, in hepatocellular carcinoma cancer cells. J. Chemother. 2012, 24, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Veschi, S.; De Lellis, L.; Florio, R.; Lanuti, P.; Massucci, A.; Tinari, N.; De Tursi, M.; di Sebastiano, P.; Marchisio, M.; Natoli, C.; et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 236. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Du, L.; Pham, P.; Zhu, B.; Jiang, S. Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell cycle arrest in human cervical cancer cells via the ROS-dependent mitochondrial pathway. Cancer Lett. 2015, 364, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Chen, R.; Chen, J.; Qi, Q.; Pan, Y.; Du, L.; Xiao, G.; Jiang, S. Combining metformin and nelfinavir exhibits synergistic effects against the growth of human cervical cancer cells and xenograft in nude mice. Sci. Rep. 2017, 7, 43373. [Google Scholar] [CrossRef] [Green Version]
- Bruning, A.; Burger, P.; Vogel, M.; Gingelmaier, A.; Friese, K.; Burges, A. Nelfinavir induces mitochondria protection by ERK1/2-mediated mcl-1 stabilization that can be overcome by sorafenib. Investig. New Drugs 2010, 28, 535–542. [Google Scholar] [CrossRef]
- Cande, C.; Cecconi, F.; Dessen, P.; Kroemer, G. Apoptosis-inducing factor (AIF): Key to the conserved caspase-independent pathways of cell death? J. Cell Sci. 2002, 115, 4727–4734. [Google Scholar] [CrossRef] [Green Version]
- Bono, C.; Karlin, L.; Harel, S.; Mouly, E.; Labaume, S.; Galicier, L.; Apcher, S.; Sauvageon, H.; Fermand, J.P.; Bories, J.C.; et al. The human immunodeficiency virus-1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica 2012, 97, 1101–1109. [Google Scholar] [CrossRef]
- Kushchayeva, Y.; Jensen, K.; Recupero, A.; Costello, J.; Patel, A.; Klubo-Gwiezdzinska, J.; Boyle, L.; Burman, K.; Vasko, V. The HIV protease inhibitor nelfinavir down-regulates RET signaling and induces apoptosis in medullary thyroid cancer cells. J. Clinl. Endocrinol. Metabol. 2014, 99, E734–E745. [Google Scholar] [CrossRef] [Green Version]
- Bruning, A.; Friese, K.; Burges, A.; Mylonas, I. Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Res. 2010, 12, R45. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Thomas, S.; Golden, E.B.; Gaffney, K.J.; Hofman, F.M.; Chen, T.C.; Louie, S.G.; Petasis, N.A.; Schonthal, A.H. Enhanced killing of chemo-resistant breast cancer cells via controlled aggravation of ER stress. Cancer Lett. 2009, 282, 87–97. [Google Scholar] [CrossRef]
- Thomas, S.; Sharma, N.; Golden, E.B.; Cho, H.; Agarwal, P.; Gaffney, K.J.; Petasis, N.A.; Chen, T.C.; Hofman, F.M.; Louie, S.G.; et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012, 325, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Delaney, J.R.; Patel, C.B.; Storgard, R.; Stupack, D.G. Nelfinavir is effective against human cervical cancer cells in vivo: A potential treatment modality in resource-limited settings. Drug Des. Devel. Ther. 2016, 10, 1837–1846. [Google Scholar] [PubMed] [Green Version]
- Bruning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, V.; Riemer, J.; Narendran, A. The HIV protease inhibitor, nelfinavir, as a novel therapeutic approach for the treatment of refractory pediatric leukemia. Onco. Targets Ther. 2017, 10, 2581–2593. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Meng, Q.; Sun, Y.; Wang, C.; Huo, X.; Liu, Z.; Sun, P.; Sun, H.; Ma, X.; Liu, K. Targeting P-Glycoprotein: Nelfinavir Reverses Adriamycin Resistance in K562/ADR Cells. Cell Physiol. Biochem. 2018, 51, 1616–1631. [Google Scholar] [CrossRef]
- Mathur, A.; Abd Elmageed, Z.Y.; Liu, X.; Kostochka, M.L.; Zhang, H.; Abdel-Mageed, A.B.; Mondal, D. Subverting ER-stress towards apoptosis by nelfinavir and curcumin coexposure augments docetaxel efficacy in castration resistant prostate cancer cells. PLoS ONE 2014, 9, e103109. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Takeuchi, T.; Adachi, Y.; Kobayashi, M.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J. Cancer 2006, 95, 1653–1662. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ikezoe, T.; Takeuchi, T.; Adachi, Y.; Ohtsuki, Y.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human prostate cancer LNCaP cells in vitro and in vivo in conjunction with blockade of androgen receptor STAT3 and AKT signaling. Cancer Sci. 2005, 96, 425–433. [Google Scholar] [CrossRef]
- Vandewynckel, Y.P.; Coucke, C.; Laukens, D.; Devisscher, L.; Paridaens, A.; Bogaerts, E.; Vandierendonck, A.; Raevens, S.; Verhelst, X.; Van Steenkiste, C.; et al. Next-generation proteasome inhibitor oprozomib synergizes with modulators of the unfolded protein response to suppress hepatocellular carcinoma. Oncotarget 2016, 7, 34988–35000. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Samuleson, C.G.; Su, S.; Chen, T.C. Nelfinavir potentiation of imatinib cytotoxicity in meningioma cells via survivin inhibition. Neurosurg. Focus 2007, 23, E9. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ye, J.; Alonso-Basanta, M.; Hahn, S.M.; Koumenis, C.; Dorsey, J.F. Modulation of CCAAT/enhancer binding protein homologous protein (CHOP)-dependent DR5 expression by nelfinavir sensitizes glioblastoma multiforme cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 2011, 286, 29408–29416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruning, A.; Vogel, M.; Burger, P.; Rahmeh, M.; Gingelmaier, A.; Friese, K.; Lenhard, M.; Burges, A. Nelfinavir induces TRAIL receptor upregulation in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2008, 377, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta 2014, 1843, 2143–2149. [Google Scholar] [CrossRef] [Green Version]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, S.; Gills, J.J.; Mercado-Matos, J.R.; Lopiccolo, J.; Wilson, W., 3rd; Hollander, M.C.; Dennis, P.A. Synergistic effects of nelfinavir and bortezomib on proteotoxic death of NSCLC and multiple myeloma cells. Cell Death Dis. 2012, 3, e353. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Gills, J.J.; Ballas, M.S.; Bernstein, W.B.; Komiya, T.; Dechowdhury, R.; Morrow, B.; Root, H.; Chun, G.; Helsabeck, C.; et al. A phase I trial of the HIV protease inhibitor nelfinavir in adults with solid tumors. Oncotarget 2014, 5, 8161–8172. [Google Scholar] [CrossRef] [Green Version]
- Pyrko, P.; Kardosh, A.; Wang, W.; Xiong, W.; Schonthal, A.H.; Chen, T.C. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007, 67, 10920–10928. [Google Scholar] [CrossRef] [Green Version]
- Mahameed, M.; Boukeileh, S.; Obiedat, A.; Darawshi, O.; Dipta, P.; Rimon, A.; McLennan, G.; Fassler, R.; Reichmann, D.; Karni, R.; et al. Pharmacological induction of selective endoplasmic reticulum retention as a strategy for cancer therapy. Nat. Commun. 2020, 11, 1304. [Google Scholar] [CrossRef]
- Chakravarty, G.; Mathur, A.; Mallade, P.; Gerlach, S.; Willis, J.; Datta, A.; Srivastav, S.; Abdel-Mageed, A.B.; Mondal, D. Nelfinavir targets multiple drug resistance mechanisms to increase the efficacy of doxorubicin in MCF-7/Dox breast cancer cells. Biochimie 2016, 124, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.; Fousek, K.; Jiang, C.; Guo, S.; Synold, T.; Xi, B.; Shih, C.C.; Chow, W.A. Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin. Cancer Res. 2011, 17, 1796–1806. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.; Fousek, K.; Chow, W.A. Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. FEBS J. 2012, 279, 2399–2411. [Google Scholar] [CrossRef]
- Sakakura, Y.; Shimano, H.; Sone, H.; Takahashi, A.; Inoue, N.; Toyoshima, H.; Suzuki, S.; Yamada, N. Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem. Biophys. Res. Commun. 2001, 286, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Ye, J.; Rawson, R.B.; Goldstein, J.L. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 2000, 100, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.; Su, L.; Yuan, Y.C.; Li, H.; Chow, W.A. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci. Rep. 2015, 5, 9698. [Google Scholar] [CrossRef] [PubMed]
- De Gassart, A.; Martinon, F. Translating the anticancer properties of eEF2K. Cell Cycle. 2017, 16, 299–300. [Google Scholar] [CrossRef] [Green Version]
- De Gassart, A.; Demaria, O.; Panes, R.; Zaffalon, L.; Ryazanov, A.G.; Gilliet, M.; Martinon, F. Pharmacological eEF2K activation promotes cell death and inhibits cancer progression. EMBO Rep. 2016, 17, 1471–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gassart, A.; Bujisic, B.; Zaffalon, L.; Decosterd, L.A.; Di Micco, A.; Frera, G.; Tallant, R.; Martinon, F. An inhibitor of HIV-1 protease modulates constitutive eIF2α dephosphorylation to trigger a specific integrated stress response. Proc. Natl. Acad. Sci. USA 2016, 113, E117–E126. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.E.; Hunt, D.K.; Wiltshire, M.; Herbert, T.P.; Sampson, J.R.; Errington, R.J.; Davies, D.M.; Tee, A.R. Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol. Oncol. 2015, 9, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.E.; Dunlop, E.A.; Seifan, S.; McCann, H.D.; Hay, T.; Parfitt, G.J.; Jones, A.T.; Giles, P.J.; Shen, M.H.; Sampson, J.R.; et al. Loss of tuberous sclerosis complex 2 sensitizes tumors to nelfinavir-bortezomib therapy to intensify endoplasmic reticulum stress-induced cell death. Oncogene 2018, 37, 5913–5925. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Johnson, C.E.; Wiltshire, M.; Errington, R.J.; Tee, A.R. Targeting protein homeostasis with nelfinavir/salinomycin dual therapy effectively induces death of mTORC1 hyperactive cells. Oncotarget 2017, 8, 48711–48724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, H.D.; Johnson, C.E.; Errington, R.J.; Davies, D.M.; Dunlop, E.A.; Tee, A.R. Energy Stress-Mediated Cytotoxicity in Tuberous Sclerosis Complex 2-Deficient Cells with Nelfinavir and Mefloquine Treatment. Cancers 2018, 10, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, M.; Bader, J.; Overkleeft, H.; Driessen, C. Nelfinavir augments proteasome inhibition by bortezomib in myeloma cells and overcomes bortezomib and carfilzomib resistance. Blood Cancer J. 2013, 3, e103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, M.; Muller-Ide, H.; Ruckrich, T.; Bader, J.; Overkleeft, H.; Driessen, C. Ritonavir, nelfinavir, saquinavir and lopinavir induce proteotoxic stress in acute myeloid leukemia cells and sensitize them for proteasome inhibitor treatment at low micromolar drug concentrations. Leuk Res. 2014, 38, 383–392. [Google Scholar] [CrossRef]
- Driessen, C.; Kraus, M.; Joerger, M.; Rosing, H.; Bader, J.; Hitz, F.; Berset, C.; Xyrafas, A.; Hawle, H.; Berthod, G.; et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: A phase I trial (SAKK 65/08). Haematologica 2016, 101, 346–355. [Google Scholar] [CrossRef]
- Hitz, F.; Kraus, M.; Pabst, T.; Hess, D.; Besse, L.; Silzle, T.; Novak, U.; Seipel, K.; Rondeau, S.; Studeli, S.; et al. Nelfinavir and lenalidomide/dexamethasone in patients with lenalidomide-refractory multiple myeloma. A phase I/II Trial (SAKK 39/10). Blood Cancer J. 2019, 9, 70. [Google Scholar] [CrossRef]
- Mahoney, E.; Maddocks, K.; Flynn, J.; Jones, J.; Cole, S.L.; Zhang, X.; Byrd, J.C.; Johnson, A.J. Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: A new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk Lymphoma. 2013, 54, 2685–2692. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Gills, J.J.; Lopiccolo, J.; Dennis, P.A. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 2008, 4, 107–109. [Google Scholar] [CrossRef] [Green Version]
- Escalante, A.M.; McGrath, R.T.; Karolak, M.R.; Dorr, R.T.; Lynch, R.M.; Landowski, T.H. Preventing the autophagic survival response by inhibition of calpain enhances the cytotoxic activity of bortezomib in vitro and in vivo. Cancer Chemother. Pharmacol. 2013, 71, 1567–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demarchi, F.; Bertoli, C.; Copetti, T.; Tanida, I.; Brancolini, C.; Eskelinen, E.-L.; Schneider, C. Calpain is required for macroautophagy in mammalian cells. J. Cell Biol. 2006, 175, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navon, A.; Ciechanover, A. The 26 S proteasome: From basic mechanisms to drug targeting. J. Biol. Chem. 2009, 284, 33713–33718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [Green Version]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.S.; Rao, R.; Beebe, K.; Neckers, L.; Han, I.; Nahta, R.; Liu, J.O. Selective inhibition of HER2-positive breast cancer cells by the HIV protease inhibitor nelfinavir. J. Natl. Cancer Inst. 2012, 104, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Li, B.; Cerniglia, G.J.; Ahmed, M.S.; Hahn, S.M.; Maity, A. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia 2007, 9, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Pajonk, F.; Himmelsbach, J.; Riess, K.; Sommer, A.; McBride, W.H. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res. 2002, 62, 5230–5235. [Google Scholar]
- Piccinini, M.; Rinaudo, M.T.; Anselmino, A.; Buccinnà, B.; Ramondetti, C.; Dematteis, A.; Ricotti, E.; Palmisano, L.; Mostert, M.; Tovo, P.A. The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antivir. Ther. 2005, 10, 215–223. [Google Scholar] [PubMed]
- Fassmannová, D.; Sedlák, F.; Sedláček, J.; Špička, I.; Grantz Šašková, K. Nelfinavir Inhibits the TCF11/Nrf1-Mediated proteasome recovery pathway in multiple myeloma. Cancers 2020, 12, 1065. [Google Scholar] [CrossRef] [PubMed]
- Alfano, L.; Guida, T.; Provitera, L.; Vecchio, G.; Billaud, M.; Santoro, M.; Carlomagno, F. RET is a heat shock protein 90 (HSP90) client protein and is knocked down upon HSP90 pharmacological block. J. Clin. Endocrinol. Metab. 2010, 95, 3552–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlomagno, F.; Guida, T.; Anaganti, S.; Vecchio, G.; Fusco, A.; Ryan, A.J.; Billaud, M.; Santoro, M. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004, 23, 6056–6063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrich, A.M.; Leshchenko, V.; Kuo, P.Y.; Xia, B.; Thirukonda, V.K.; Ulahannan, N.; Gordon, S.; Fazzari, M.J.; Ye, B.H.; Sparano, J.A.; et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clin. Cancer Res. 2012, 18, 2534–2544. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; McKenna, W.G.; Muschel, R.J. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Res. 2005, 65, 8256–8265. [Google Scholar] [CrossRef] [Green Version]
- Goda, J.S.; Pachpor, T.; Basu, T.; Chopra, S.; Gota, V. Targeting the AKT pathway: Repositioning HIV protease inhibitors as radiosensitizers. Indian J. Med. Res. 2016, 143, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, E.J.; Brunner, T.B. Progress towards the use of HIV protease inhibitors in cancer therapy. Cancer Biol. Ther. 2008, 7, 636–637. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Lee, J.H.; Wilke, W.W.; Quon, H.; Smith, G.; Maity, A.; Buatti, J.M.; Spitz, D.R. Radiation response in two HPV-infected head-and-neck cancer cell lines in comparison to a non-HPV-infected cell line and relationship to signaling through AKT. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Pore, N.; Cerniglia, G.J.; Mick, R.; Georgescu, M.M.; Bernhard, E.J.; Hahn, S.M.; Gupta, A.K.; Maity, A. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res. 2007, 67, 4467–4473. [Google Scholar] [CrossRef] [Green Version]
- Kimple, R.J.; Vaseva, A.V.; Cox, A.D.; Baerman, K.M.; Calvo, B.F.; Tepper, J.E.; Shields, J.M.; Sartor, C.I. Radiosensitization of epidermal growth factor receptor/HER2-positive pancreatic cancer is mediated by inhibition of Akt independent of ras mutational status. Clin. Cancer Res. 2010, 16, 912–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuneo, K.C.; Tu, T.; Geng, L.; Fu, A.; Hallahan, D.E.; Willey, C.D. HIV protease inhibitors enhance the efficacy of irradiation. Cancer Res. 2007, 67, 4886–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Jiang, Z.; Bernhard, E.J.; Evans, S.M.; Koch, C.J.; Hahn, S.M.; Maity, A. Nelfinavir down-regulates hypoxia-inducible factor 1alpha and VEGF expression and increases tumor oxygenation: Implications for radiotherapy. Cancer Res. 2006, 66, 9252–9259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pore, N.; Gupta, A.K.; Cerniglia, G.J.; Maity, A. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 2006, 8, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; See, A.P.; Aziz, K.; Thiyagarajan, S.; Salih, T.; Gajula, R.P.; Armour, M.; Phallen, J.; Terezakis, S.; Kleinberg, L.; et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biol. Ther. 2011, 12, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Plastaras, J.P.; Vapiwala, N.; Ahmed, M.S.; Gudonis, D.; Cerniglia, G.J.; Feldman, M.D.; Frank, I.; Gupta, A.K. Validation and toxicity of PI3K/Akt pathway inhibition by HIV protease inhibitors in humans. Cancer Biol. Ther. 2008, 7, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Brunner, T.B.; Geiger, M.; Grabenbauer, G.G.; Lang-Welzenbach, M.; Mantoni, T.S.; Cavallaro, A.; Sauer, R.; Hohenberger, W.; McKenna, W.G. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 2008, 26, 2699–2706. [Google Scholar] [CrossRef]
- Gupta, A.K.; Wilke, W.W.; Taylor, E.N.; Bodeker, K.L.; Hoffman, H.T.; Milhem, M.M.; Buatti, J.M.; Robinson, R.A. Signaling pathways in adenoid cystic cancers: Implications for treatment. Cancer Biol. Ther. 2009, 8, 1947–1951. [Google Scholar] [CrossRef] [Green Version]
- Hoover, A.C.; Milhem, M.M.; Anderson, C.M.; Sun, W.; Smith, B.J.; Hoffman, H.T.; Buatti, J.M. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: Results of a phase II clinical trial. Head Neck 2015, 37, 722–726. [Google Scholar] [CrossRef]
- Liebscher, S.; Koi, L.; Lock, S.; Muders, M.H.; Krause, M. The HIV protease and PI3K/Akt inhibitor nelfinavir does not improve the curative effect of fractionated irradiation in PC-3 prostate cancer in vitro and in vivo. Clin. Transl. Radiat. Oncol. 2017, 2, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Bruning, A. Targeting the off-targets: A computational bioinformatics approach to understanding the polypharmacology of nelfinavir. Expert Rev. Clin. Pharmacol. 2011, 4, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Saito, T.; Bandobashi, K.; Yang, Y.; Koeffler, H.P.; Taguchi, H. HIV-1 protease inhibitor induces growth arrest and apoptosis of human multiple myeloma cells via inactivation of signal transducer and activator of transcription 3 and extracellular signal-regulated kinase 1/2. Mol. Cancer Ther. 2004, 3, 473–479. [Google Scholar] [PubMed]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxid. Med. Cell Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef] [Green Version]
- Kushchayeva, Y.; Jensen, K.; Burman, K.D.; Vasko, V. Repositioning therapy for thyroid cancer: New insights on established medications. Endocr. Relat. Cancer 2014, 21, R183–R194. [Google Scholar] [CrossRef]
- Xia, C.; He, Z.; Liang, S.; Chen, R.; Xu, W.; Yang, J.; Xiao, G.; Jiang, S. Metformin combined with nelfinavir induces SIRT3/mROS-dependent autophagy in human cervical cancer cells and xenograft in nude mice. Eur. J. Pharmacol. 2019, 848, 62–69. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef] [Green Version]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Qayum, N.; Im, J.; Stratford, M.R.; Bernhard, E.J.; McKenna, W.G.; Muschel, R.J. Modulation of the tumor microvasculature by phosphoinositide-3 kinase inhibition increases doxorubicin delivery in vivo. Clin. Cancer Res. 2012, 18, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Bourlier, V.; Zakaroff-Girard, A.; De Barros, S.; Pizzacalla, C.; de Saint Front, V.D.; Lafontan, M.; Bouloumie, A.; Galitzky, J. Protease inhibitor treatments reveal specific involvement of matrix metalloproteinase-9 in human adipocyte differentiation. J. Pharmacol. Exp. Ther. 2005, 312, 1272–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, R.E.; Halatsch, M.E. Matrix metalloproteinase-2 and -9 in glioblastoma: A trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch. Med. Res. 2012, 43, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Al-Assar, O.; Bittner, M.I.; Lunardi, S.; Stratford, M.R.; McKenna, W.G.; Brunner, T.B. The radiosensitizing effects of Nelfinavir on pancreatic cancer with and without pancreatic stellate cells. Radiother. Oncol. 2016, 119, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Darini, C.Y.; Martin, P.; Azoulay, S.; Drici, M.D.; Hofman, P.; Obba, S.; Dani, C.; Ladoux, A. Targeting cancer stem cells expressing an embryonic signature with anti-proteases to decrease their tumor potential. Cell Death. Dis. 2013, 4, e706. [Google Scholar] [CrossRef] [Green Version]
- Giardino Torchia, M.L.; Ciaglia, E.; Masci, A.M.; Vitiello, L.; Fogli, M.; la Sala, A.; Mavilio, D.; Racioppi, L. Dendritic cells/natural killer cross-talk: A novel target for human immunodeficiency virus type-1 protease inhibitors. PLoS ONE 2010, 5, e11052. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Ghatak, S.; Toole, B.P. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 2005, 280, 20310–20315. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Park, Y.J.; Lee, B.M.; Yoon, S. Co-treatment With HIV Protease Inhibitor Nelfinavir Greatly Increases Late-phase Apoptosis of Drug-resistant KBV20C Cancer Cells Independently of P-Glycoprotein Inhibition. Anticancer Res. 2019, 39, 3757–3765. [Google Scholar] [CrossRef]
- Lucia, M.B.; Anu, R.; Handley, M.; Gillet, J.P.; Wu, C.P.; De Donatis, G.M.; Cauda, R.; Gottesman, M.M. Exposure to HIV-protease inhibitors selects for increased expression of P-glycoprotein (ABCB1) in Kaposi’s sarcoma cells. Br. J. Cancer 2011, 105, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Perloff, M.D.; von Moltke, L.L.; Fahey, J.M.; Daily, J.P.; Greenblatt, D.J. Induction of P-glycoprotein expression by HIV protease inhibitors in cell culture. AIDS 2000, 14, 1287–1289. [Google Scholar] [CrossRef]
- Gupta, A.; Zhang, Y.; Unadkat, J.D.; Mao, Q. HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J. Pharmacol. Exp. Ther. 2004, 310, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, Y.; Takenaka, K.; Sparreboom, A.; Cheepala, S.B.; Wu, C.P.; Ekins, S.; Ambudkar, S.V.; Schuetz, J.D. Human immunodeficiency virus protease inhibitors interact with ATP binding cassette transporter 4/multidrug resistance protein 4: A basis for unanticipated enhanced cytotoxicity. Mol. Pharmacol. 2013, 84, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengan, R.; Mick, R.; Pryma, D.; Rosen, M.A.; Lin, L.L.; Maity, A.M.; Evans, T.L.; Stevenson, J.P.; Langer, C.J.; Kucharczuk, J.; et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: A report of toxicities and clinical response. J. Thorac. Oncol. 2012, 7, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengan, R.; Mick, R.; Pryma, D.A.; Lin, L.L.; Christodouleas, J.; Plastaras, J.P.; Simone, C.B., 2nd; Gupta, A.K.; Evans, T.L.; Stevenson, J.P.; et al. Clinical Outcomes of the HIV Protease Inhibitor Nelfinavir With Concurrent Chemoradiotherapy for Unresectable Stage IIIA/IIIB Non-Small Cell Lung Cancer: A Phase 1/2 Trial. JAMA Oncol. 2019, 5, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Fokas, E.; Dutton, S.J.; Patel, N.; Hawkins, M.A.; Eccles, C.; Chu, K.Y.; Durrant, L.; Abraham, A.G.; Partridge, M.; et al. ARCII: A phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother. Oncol. 2016, 119, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Strauss, V.Y.; Shaw, R.; Virdee, P.S.; Hurt, C.N.; Ward, E.; Tranter, B.; Patel, N.; Bridgewater, J.; Parsons, P.; Radhakrishna, G.; et al. Study protocol: A multi-centre randomised study of induction chemotherapy followed by capecitabine +/− nelfinavir with high- or standard-dose radiotherapy for locally advanced pancreatic cancer (SCALOP-2). BMC Cancer 2019, 19, 121. [Google Scholar] [CrossRef]
- Lin, C.; Verma, V.; Ly, Q.P.; Lazenby, A.; Sasson, A.; Schwarz, J.K.; Meza, J.L.; Are, C.; Li, S.; Wang, S.; et al. Phase I trial of concurrent stereotactic body radiotherapy and nelfinavir for locally advanced borderline or unresectable pancreatic adenocarcinoma. Radiother. Oncol. 2019, 132, 55–62. [Google Scholar] [CrossRef]
- Lin, C.; Verma, V.; Lazenby, A.; Ly, Q.P.; Berim, L.D.; Schwarz, J.K.; Madiyalakan, M.; Nicodemus, C.F.; Hollingsworth, M.A.; Meza, J.L.; et al. Phase I/II Trial of Neoadjuvant Oregovomab-based Chemoimmunotherapy Followed by Stereotactic Body Radiotherapy and Nelfinavir For Locally Advanced Pancreatic Adenocarcinoma. Am. J. Clin. Oncol. 2019, 42, 755–760. [Google Scholar] [CrossRef]
- Pan, J.; Mott, M.; Xi, B.; Hepner, E.; Guan, M.; Fousek, K.; Magnusson, R.; Tinsley, R.; Valdes, F.; Frankel, P.; et al. Phase I study of nelfinavir in liposarcoma. Cancer Chemother. Pharmacol. 2012, 70, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Hill, E.J.; Roberts, C.; Franklin, J.M.; Enescu, M.; West, N.; MacGregor, T.P.; Chu, K.Y.; Boyle, L.; Blesing, C.; Wang, L.M.; et al. Clinical Trial of Oral Nelfinavir before and during Radiation Therapy for Advanced Rectal Cancer. Clin. Cancer Res. 2016, 22, 1922–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijsen, J.; Lammering, G.; Jansen, R.L.; Beets, G.L.; Wals, J.; Sosef, M.; Den Boer, M.O.; Leijtens, J.; Riedl, R.G.; Theys, J.; et al. Phase I trial of the combination of the Akt inhibitor nelfinavir and chemoradiation for locally advanced rectal cancer. Radiother. Oncol. 2013, 107, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, W.B.; Dennis, P.A. Repositioning HIV protease inhibitors as cancer therapeutics. Curr. Opin. HIV AIDS 2008, 3, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Basanta, M.; Fang, P.; Maity, A.; Hahn, S.M.; Lustig, R.A.; Dorsey, J.F. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J. Neurooncol. 2014, 116, 365–372. [Google Scholar] [CrossRef]
- Kattel, K.; Evande, R.; Tan, C.; Mondal, G.; Grem, J.L.; Mahato, R.I. Impact of CYP2C19 polymorphism on the pharmacokinetics of nelfinavir in patients with pancreatic cancer. Br. J. Clin. Pharmacol. 2015, 80, 267–275. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Publications (et al.) | Cancer Type | Animal Background | Cells and Method of Xenograft | Dosing of Nelfinavir ± Co-Treatment | Time | Main Result(s) |
|---|---|---|---|---|---|---|
| Al Assar, 2016 [123] | Pancreatic cancer | Female nude mice | PSN-1, SC 1, Flank | 20 mg/kg, IP 2 ± RT 3 3.5 Gy | 20 d | Overcoming radioprotective effect of pancreatic stellate cells |
| Bono, 2011 [37] | Multiple myeloma | NOD/SCID 4 mice | U266-Luc 5, SC, Flank | 75 mg/kg, IP | 21 d | Reduced tumor burden |
| Chakravarty, 2016 [60] | Breast cancer | Female athymic nude BALB/c mice | MCF-Dox 6, 4th inguinal mammary gland (orthotopic) | 20 mg/kg, IP ± Dox 2 mg/kg | 6 w | Reduced tumor growth and p-AKT |
| Cuneo, 2007 [102] | Lung cancer | C7/BL6 mice | Lewis lung carcinoma, SC, hind limb | 30 mg/kg, oral ± RT 2 Gy | 3–5 d | Reduced vascular density and angiogenesis |
| Davis, 2016 [42] | Cervical cancer | Female athymic nude mice | ME-180, ME-180 CPR 7, SC, alternate flanks | 250 mg/kg/d, gastric gavage | 21 d | Reduced tumor growth of both cisplatin sensitive and resistant cells |
| De Gassart, 2016 [67] | Spontaneous | Immuno-compromised AGR 129 mice | eEF2K 8 WT 9 HRasV12, eEF2k−/− HRasV12, SC. Alternate flanks | 100 mg/kg, IP | Tumor growth inhibition in response to nelfinavir in eEF2K WT mice but not in eEF2K-deficient mice | |
| Escalante, 2013 [81] | Multiple myeloma | SCID mice | MM.1S, SC | 50 mg/kg, oral gavage ± BZ 10 1 mg/kg, IV tail vein | Until 10% wt 11 loss | Complete tumor regression in combination group |
| Gills, 2007 [19] | Lung cancer | Balb/cAnCr nu/nu mice, athymic nude mice | H157, A548; SC, shoulder and rear flanks | 50–100 mg/kg, IP; or 100 mg/kg gastric gavage | 10–20 d | Tumor growth delay, ER stress, autophagy |
| Guan, 2011 [61] | Liposarcoma | SCID mice | Lisa-2, SC, heterotopic model | 500 mg/kg/d, diet | 41 d | Reduced tumor growth |
| Gupta, 2007 [50] | Meningioma | Male athymic nu/nu mice | IOMM-Lee, SC, right flank | 150 mg/kg/d, oral ± Imatinib 100 mg/kg/d | 23 d | Combined treatment caused tumor growth reduction, ER stress, apoptosis and reduced level of survivin |
| Gupta, 2005 [96] | Head and neck cancer, bladder cancer | NCr-nu/nu mice | SQ20B (EGFR mutated), T24 (HRas mutated), SC, hind flank | 0.6 mg/day, continuous release pellets ± RT 6–8 Gy | Time to reach 1000 mm3 | Combined treatment caused tumor regrowth delay |
| Jiang, 2007 [100] | Glioblastoma | Female NCr-nu/nu mice | U87MG (PTEN deficient), SC, flank | 79 mg/kg/day, diet ± RT 6 Gy | Time to reach 1000 mm3 | Combined treatment caused tumor growth delay; nelfinavir reduced p-Akt |
| Johnson, 2018 [70] | Tuberous sclerosis complex | NOD/SCID female mice | ELT3-V3 (Tsc2−/−), SC, right flank | 30–50 mg/kg, IP ± BZ 0.3–0.5 mg/kg | 17 d | Combined treatment caused tumor growth reduction, ER stress, apoptosis |
| Kawabata, 2012 [56] | NSCLC 12, multiple myeloma | Athymic NCr nu/nu mice | H157, RPMI8226, SC, both rear flanks | 50 mg/kg, IP ± BZ 0.5 mg/kg | 11–17 d | Combined treatment caused tumor growth reduction, ER stress, apoptosis |
| Kimple, 2010 [101] | Pancreatic cancer | Athymic BALB/c nude mice | Capan-2, SC, flanks | 150 mg/kg, Oral gavage ± RT 200 cGy/day | 10 d | Combined treatment caused tumor growth reduction; nelfinavir reduced p-Akt |
| Mathur, 2014 [46] | Castration-resistant prostate cancer | Athymic nude mice | C4–2B, SC | DTX 13 (10 mg/kg), ± [nelfinavir (20 mg/kg) and curcumin (100 mg/kg)] | 4 w | Triple combination caused tumor growth delay and apoptosis |
| Okubo, 2018 [29] | Renal cancer | BALB/c male nude mice | Caki-2, SC | 25 mg/kg, IP ± PAN 14 (2 mg/kg) | 11 d | Combined treatment caused tumor growth reduction, ER stress, apoptosis and histone acetylation |
| Pore, 2006 [103] | Lung cancer, head neck squamous cell cancer | BALB/c NCr nu/nu mice | A549, SQ20B, SC, flank | 79 mg/kg/d, diet; ± RT 8 Gy | Time to reach 1000 mm3 | Combined treatment reduced tumor growth; nelfinavir reduced angiogenesis and VEGF 15 |
| Pore, 2006 [104] | Glioblastoma | BALB/c NCr nu/nu mice | U87, SC | 40 mg/kg/d; diet | 5 d | Reduced angiogenesis |
| Pyrko, 2007 [58] | Glioblastoma | Male athymic nu/nu mice | U87, SC | 40 mg/kg/d (short-term), 120 mg/kg/d (long-term); gastric gavage | 96 h (short- term), 6 w (long-term) | Tumor growth reduction, ER stress, apoptosis |
| Qayum, 2009 [120] | Fibrosarcoma, Laryngeal cancer | SCID mice | HT1080, SQ20B, SC, hind leg | 20 mg/kg, IP | 2 w | Reduced tumor hypoxia, increased tumor blood flow, normalized tumor vascular morphology |
| Shim, 2012 [88] | Breast cancer | BABL/c NCr nu/nu mice | HER2 16 positive: HCC1954, BT474; HER2 negative: HCC1937, MDA-MB-231, SC | 25 mg/kg, IP; 40 mg/kg, oral | 30 d | Nelfinavir selectively inhibited the growth of HER2-positive tumors and decreased expression of HER2 |
| Smith, 2016 [113] | Melanoma | Nude mice | A375, M249-R4, SC | 25 mg/kg/qd, oral gavage ± MEKi 17 (25 mg/mg/qd) or BRAFi 18 (25 mg/kg/qd) | 21–33 d | Combined treatment caused reduction in tumor growth and expression of PAX and MITF 19 |
| Thomas, 2012 [41] | Breast cancer | Athymic mice | MDA-MB-468 (TNBC 20), MCF-7, SC, flank | 5 mg/kg/d, gavage ± Celecoxib (2 mg/kg/d) ± CQ 21 (10 mg/kg/d) | 3–5 d | Triple combination caused tumor growth reduction, ER stress and apoptosis |
| Vandewynckel, 2016 [49] | Hepatocellular carcinoma | WT 129s2/SvPasCrl mice injected with DEN 19 (orthotopic model); Athymic nude mice: Foxn1nu/foxn1nu (Xenograft model) | HepG2, SC, right flank | OZ 22 (30 mg/kg/d), intragastric ± nelfinavir (250 mg/kg/d), IP or salubrinal (1 mg/kg/d), IP | 4 w | Decreased tumor growth and increased apoptosis in both orthotopic and xenograft models |
| Xia, 2017 [34] | Cervical cancer | Female BALB/c nude mice | SiHa, SC, left flank | 0.4 mg/kg/d, IP ± metformin 100 mg/kg/d | 24 d | Reduced tumor growth and PI3K 23 expression and increased expression of p53 and p21 in response to either monotherapy or combined therapy |
| Xia, 2019 [117] | Cervical cancer | Female BABLB/c nude mice | SiHa, SC, left flank | 0.4 mg/kg/d, IP ± metformin 100 mg/kg/d | 25 d | Combined treatment caused tumor growth reduction and enhanced level of sirtuin-3 and MICA 24, suggesting NK 25 cell-mediated lysis |
| Xiang, 2015 [33] | Cervical cancer | BALB/c nude mice | HeLa, SC, back | 1 mg/mouse, IP | 20 d | Tumor growth reduction, increased apoptosis, nuclear localization of AIF 26 |
| Yang, 2006 [47] | NSCLC | BALB/c triple-deficient male nude mice | NCI-H460, SC, bilateral | 60 mg/kg, oral gavage | 3 w | Tumor growth reduction, apoptosis |
| Yang, 2005 [48] | Prostate cancer | Immunodeficient BALB/c nude mice | LNCaP, SC, bilateral | 60 mg/kg, oral gavage | 3 w | Tumor growth reduction, reduced serum level of PSA 27, increased fibrosis and inflammatory cells |
| Zeng, 2011 [105] | Pituitary adenoma | Female nude mice | GH3, SC, right flank | 5 μM, oral gavage ± RT 6 Gy | Until tumor size 4× | Tumor growth reduction, reduced phospho-S6 |
| NCT Number | Phase | Cancer Type | Concurrent Therapy | Timeline | Status | Total Patients | Objective | Ref |
|---|---|---|---|---|---|---|---|---|
| NCT01485731 | I | Cervical cancer | Cisplatin, RT 1 | January 2012–February 2015 | C 2 | 8 | Estimate of adverse event, MTD 3 | |
| NCT00589056 | I/II | Stage III NSCLC 4 | Cisplatin, etoposide, RT | June 2007–March 2012 | C | 55 | DLT 5, MTD | [134] |
| NCT01079286 | I | Renal cancer | Temsirolimus | June 2008–May 2011 | C | 18 | PK 6, PD 7, dose escalation | |
| NCT02363829 | I | LA 8 Cervical Cancer (Stage II–VA) | Cisplatin, Pelvic RT | February 2015–February 2020 | C | 6 | Number of AE 9 | |
| NCT01086332 | I/II | Locally advanced pancreatic cancer (LAPC) | Gemcitabine, RT | May 2009–July 2015 | T 10 | 7 | DLT | |
| NCT00704600 | Colorectal cancer | Capecitabine, Preoperative RT | September 2008–July 2013 | C | 15 | DLT, MTD | [142] | |
| NCT01447589 | I/II | NSCLC | Radical radiotherapy | February 2012–October 2012 | W 11 | - | MTD, AE | |
| NCT01445106 | I | Solid tumors | _ | December 2006–May 2011 | C | 28 | MTD, DLT, PK, PD, anti-tumor response, blood markers | [57] |
| NCT01065844 | II | Adenoid cystic head and neck carcinoma | _ | October 2009–November 2017 | C | 15 | Tumor progression | [109] |
| NCT01068327 | I | Pancreatic cancer (adeno-carcinoma/Stage III) | Gemcitabine hydrochloride, leucovorin calcium, fluorouracil, RT | November 2007–February 2015 | C | 46 | DLT, MTD, evaluate surgical resection rate, pathological and radiological response | [137] |
| NCT04169763 | I | Vulvar cancer (Stage II–IVA) | Cisplatin, external beam radiation | March 2020–December 2023 | NR 12 | 18 est. 13 | DLT, safety, dose for phase II | |
| NCT01108666 | II | Inoperable NSCLC (Stage III) | Cisplatin, paclitaxel, etoposide, proton beam radiation | March 2010–December 2018 | T | 8 | MTD, toxicity, feasibility of proton beam, clinical efficacy | |
| NCT02024009 | I/II | Non-metastatic LAPC | RT, nab-paclitaxel, gemcitabine, capecitabine, | March 2016–August 2020 | R 14 | 289 est. | OS 15, PFS 16, toxicity, QL 17 | [136] |
| NCT03422874 | I | Lymphoma | Ixazomib (MLN9708) | August 2016–August 2017 | W | _ | MTD, toxicity, PK, PD | |
| NCT01959672 | II | LAPC | Gemcitabine hydrochloride, leucovorin calcium, fluorouracil, oregovomab, RT | September 2013–December 2018 | C | 11 | Evaluate efficacy and safety of neoadjuvant chemotherapy followed by RT+ nelfinavir | [138] |
| NCT01164709 | I | Advanced hematologic malignancies | Bortezomib | July 2010–November 2013 | C | 18 | DLT, objective response, AE | [75] |
| NCT03050060 | II | Advanced melanoma, lung and kidney cancer | Atezolizumab, nivolumab, pembrolizumab, RT | June 2017–December 2021 | S 18 | 120 est | RR 19, OS, PFS, AE, immune correlative studies | |
| NCT02080416 | I | Gamma-herpes related tumor | _ | July 2014–February 2016 | T | 1 | Lytic activation of viral gene expression by nelfinavir | |
| NCT01925378 | II | Cervical dysplasia | _ | July 2012–December 2022 | R | 10 est. | Efficacy of nelfinavir | |
| NCT00791336 | II | NSCLC | RT, cisplatin, etoposide | August 2008–March 2011 | T | 1 | Pathologic complete response | |
| NCT00915694 | I | GBM 20 | Temozolomide, RT | April 2009–December 2015 | T | 15 | MTD, DLT, PFS, OS | [144] |
| NCT03256916 | III | Carcinoma cervix (Stage III) | Cisplatin, pelvic RT | January 2018–September 2025 | R | 300 | Improvement in 3 year disease-free survival | |
| NCT03829020 | I | Relapsed or refractory multiple myeloma | Bortezomib, metformin | April 2019–August 2021 | R | 36 est. | MTD, AE, hematological response | |
| NCT02188537 | II | Proteasome inhibitor-refractory myeloma | Bortezomib, dexamethasone | December 2014–April 2018 | C | 34 | RR, AE, QL | [20] |
| NCT01555281 | I/II | Multiple myeloma | Lenalidomide, dexamethasone | February 2012–December 2021 | AnR 21 | 33 | DLT, ORR 22, OS, PFS | [76] |
| NCT00233948 | I/II | Liposarcoma | _ | March 2006–July 2013 | T | 29 | DLT, MTD, ORR | |
| NCT00002185 | II | Kaposi sarcoma | _ | _ | C | 20 | Safety and efficacy | |
| NCT02207439 | II | Head and neck carcinoma | RT, platinum-based chemotherapy | July 2014–December 2020 | AnR | 28 | Determine locoregional control | |
| NCT03077451 | II | Kaposi sarcoma | _ | March 2017–October 2020 | AnR | 36 | Efficacy of dose escalation | |
| NCT00694837 | I | GBM | Temozolomide, RT | March 2009–January 2013 | C | 6 | MTD, toxicity | |
| NCT01020292 | I | Glioma | Temozolomide, RT | April 2009–December 2017 | C | 31 | MTD, DLT, PFS, OS | |
| NCT00003008 | II | Sarcoma | Indinavir, saquinavir, ritonavir, paclitaxel | June 1997–June 2006 | C | 33 | Role of HIV-PIs in plasma clearance of paclitaxel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subeha, M.R.; Telleria, C.M. The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir. Cancers 2020, 12, 3437. https://doi.org/10.3390/cancers12113437
Subeha MR, Telleria CM. The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir. Cancers. 2020; 12(11):3437. https://doi.org/10.3390/cancers12113437
Chicago/Turabian StyleSubeha, Mahbuba R., and Carlos M. Telleria. 2020. "The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir" Cancers 12, no. 11: 3437. https://doi.org/10.3390/cancers12113437