Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Epithelial–Mesenchymal Plasticity and Tumor Progression
3. Molecular Mechanisms of EMT in Cancer: Role of Targeted Protein Degradation by Ubiquitination
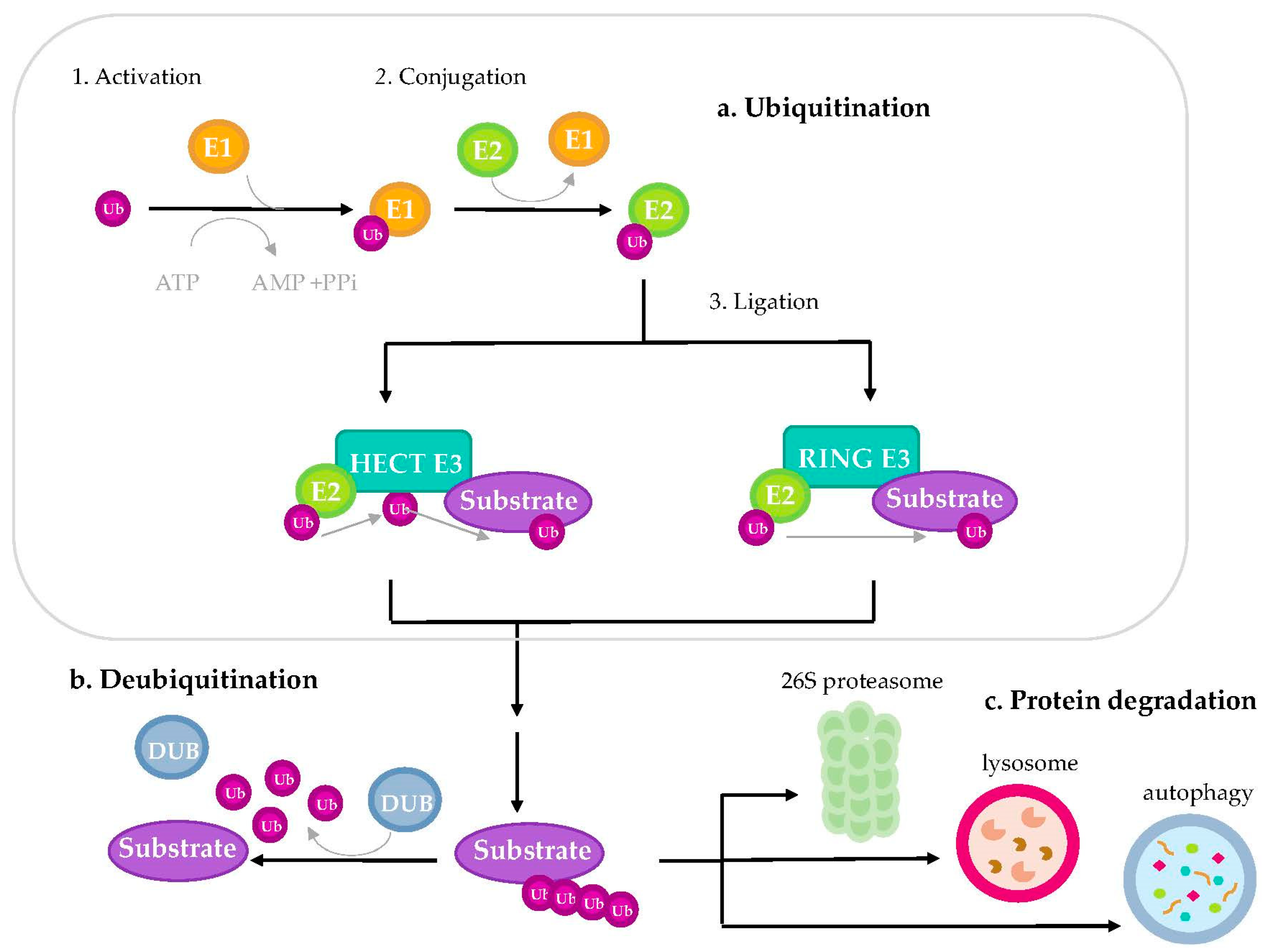
3.1. Ubiquitination Process
3.2. Classification of E3 Ubiquitin-Ligases
4. EMT Regulation by E3 Ubiquitin-Ligases
4.1. HECT-Domain E3 Ubqiuitin-Ligases
4.1.1. Smurfs
4.1.2. Hect D1
4.2. RING-Finger Domain E3 Ubqiuitin-Ligase
4.2.1. SCF Family: F-Box Proteins
4.2.2. Mdm2
4.2.3. TRIM Family
4.2.4. Rbbp6 and Ppil2
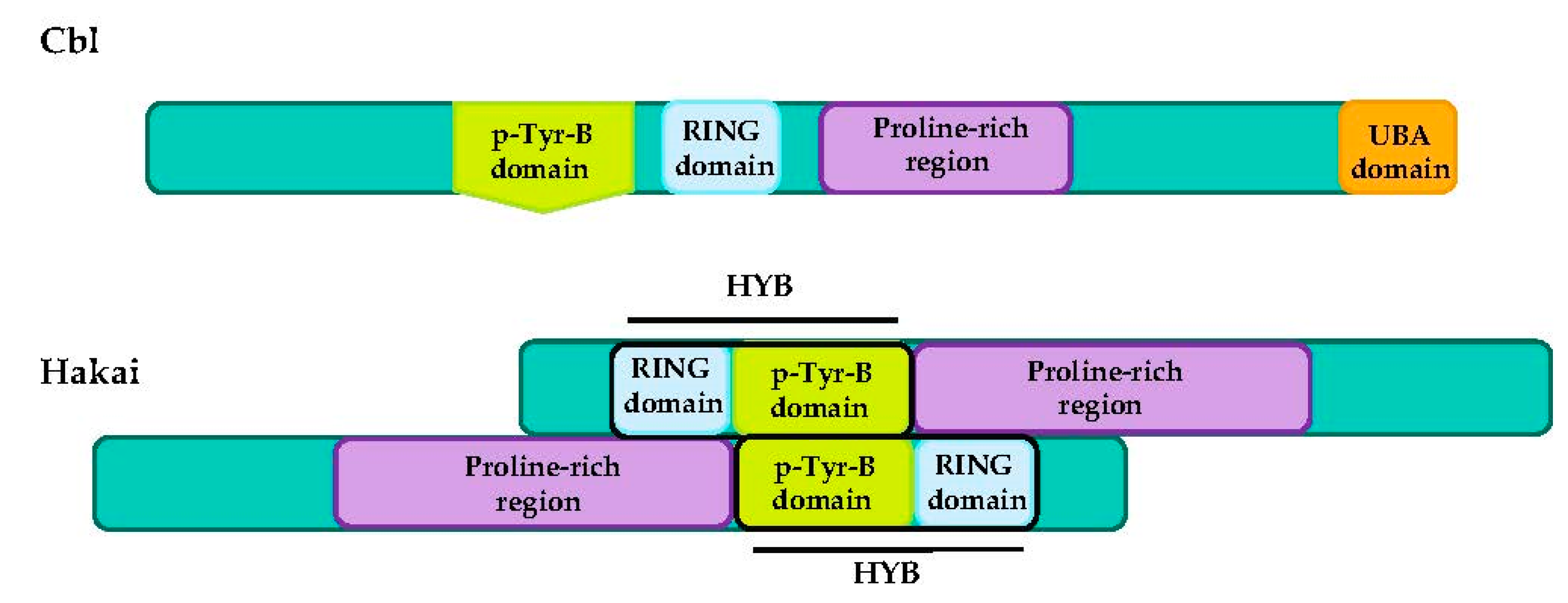
4.3. CBL Proteins
Hakai
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Steeg, P.S. Targeting metastasis. Nat. Rev. Cancer 2016, 16, 201–218. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial- mesenchymal transition to suppression of anoikis. J. Cell Sci. 2013, 126, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Perez-Moreno, M.; Jamora, C.; Fuchs, E. Sticky business: Orchestrating cellular signals at adherens junctions. Cell 2003, 112, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Blanco, M.; Castosa, R.; Concha, Á.; Valladares, M.; Calvo, L.; Figueroa, A. Clinical implications of epithelial cell plasticity in cancer progression. Cancer Lett. 2015, 366, 1–10. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Valladares, M.; Blanco, M.; Alonso, G.; Figueroa, A. Biological influence of Hakai in cancer: A 10-year review. Cancer Metastasis Rev. 2012, 31, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, ZEB and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Aparicio, L.A.; Abella, V.; Valladares, M.; Figueroa, A. Posttranscriptional regulation by RNA-binding proteins during epithelial-to-mesenchymal transition. Cell. Mol. Life Sci. 2013, 70, 4463–4477. [Google Scholar] [CrossRef] [Green Version]
- Díaz-López, A.; Moreno-Bueno, G.; Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Coppola, V.; Musumeci, M.; Patrizii, M.; Cannistraci, A.; Addario, A.; Maugeri-Saccà, M.; Biffoni, M.; Francescangeli, F.; Cordenonsi, M.; Piccolo, S.; et al. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial-mesenchymal transition. Oncogene 2013, 32, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Harvey, S.E.; Zheng, R.; Lyu, J.; Grzeskowiak, C.L.; Powell, E.; Piwnica-Worms, H.; Scott, K.L.; Cheng, C. The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, P.S. Small RNAs with big impacts. Nature 2005, 435, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Valladares-Ayerbes, M.; Reboredo, M.; Medina-Villaamil, V.; Iglesias-Díaz, P.; Lorenzo-Patiño, M.J.; Haz, M.; Santamarina, I.; Blanco, M.; Fernández-Tajes, J.; Quindós, M.; et al. Circulating miR-200c as a diagnostic and prognostic biomarker for gastric cancer. J. Transl. Med. 2012, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Moes, M.; Le Béchec, A.; Crespo, I.; Laurini, C.; Halavatyi, A.; Vetter, G.; del Sol, A.; Friederich, E. A Novel Network Integrating a miRNA-203/SNAI1 Feedback Loop which Regulates Epithelial to Mesenchymal Transition. PLoS ONE 2012, 7, e35440. [Google Scholar] [CrossRef] [Green Version]
- Abella, V.; Valladares, M.; Rodriguez, T.; Haz, M.; Blanco, M.; Tarrío, N.; Iglesias, P.; Aparicio, L.A.; Figueroa, A. miR-203 Regulates Cell Proliferation through Its Influence on Hakai Expression. PLoS ONE 2012, 7, e52568. [Google Scholar] [CrossRef] [Green Version]
- Dupre-Crochet, S.; Figueroa, A.; Hogan, C.; Ferber, E.C.; Bialucha, C.U.; Adams, J.; Richardson, E.C.N.; Fujita, Y. Casein Kinase 1 Is a Novel Negative Regulator of E-Cadherin-Based Cell-Cell Contacts. Mol. Cell. Biol. 2007, 27, 3804–3816. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, M.; Ihara, Y.; Matsuzawa, Y.; Taniguchi, N. Aberrant glycosylation of E-cadherin enhances cell-cell binding to suppress metastasis. J. Biol. Chem. 1996, 271, 13811–13815. [Google Scholar] [CrossRef] [Green Version]
- Rape, M. Post-Translational Modifications: Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef]
- Weissman, A.M.; Shabek, N.; Ciechanover, A. The predator becomes the prey: Regulating the ubiquitin system by ubiquitylation and degradation. Nat. Rev. Mol. Cell Biol. 2011, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Lipkowitz, S.; Weissman, A.M. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 2011, 11, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef]
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Polo, S.; Maspero, E. HECT E3 ligases: A tale with multiple facets. Front. Physiol. 2019, 10, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef]
- Kanemori, Y.; Uto, K.; Sagata, N. β-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc. Natl. Acad. Sci. USA 2005, 102, 6279–6284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.A.; Kaneko, T.; Li, S.S.C. Cell Regulation by Phosphotyrosine-Targeted Ubiquitin Ligases. Mol. Cell. Biol. 2015, 35, 1886–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, M.; Chow, S.Y.; Yusoff, P.; Seetharaman, J.; Ng, C.; Sinniah, S.; Koh, X.W.; Asgar, N.F.M.; Li, D.; Yim, D.; et al. Structure of a novel phosphotyrosine-binding domain in Hakai that targets E-cadherin. EMBO J. 2012, 31, 1308–1319. [Google Scholar] [CrossRef]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer Development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef]
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert Opin. Ther. Pat. 2018, 28, 919–937. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquiting-proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, S.G. Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Iglesias, O.; Casas-Pais, A.; Castosa, R.; Díaz-Díaz, A.; Roca-Lema, D.; Concha, Á.; Cortés, Á.; Gago, F.; Figueroa, A. Hakin-1, a New Specific Small-Molecule Inhibitor for the E3 Ubiquitin-Ligase Hakai, Inhibits Carcinoma Growth and Progression. Cancers 2020, 12, 1340. [Google Scholar] [CrossRef]
- Huang, X.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Wang, Y.; Fan, J.; Chen, R. Deletion of SMURF 1 represses ovarian cancer invasion and EMT by modulating the DAB2IP/AKT/Skp2 feedback loop. J. Cell. Biochem. 2019, 120, 10643–10651. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Li, Y.; Gao, Y.; Huang, S.; Li, H.; Zhu, Y. SMURF1-mediated ubiquitination of ARHGAP26 promotes ovarian cancer cell invasion and migration. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Yang, Y.A.; Anver, M.R.; Morris, N.; Wang, X.; Zhang, Y.E. Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer Res. 2009, 69, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, E.; Inoue, Y.; Komiya, S.; Horiguchi, K.; Goto, K.; Saitoh, M.; Miyazawa, K.; Koinuma, D.; Hanyu, A.; Imamura, T. Smurf2 induces ubiquitin-dependent degradation of Smurf1 to prevent migration of breast cancer cells. J. Biol. Chem. 2008, 283, 35660–35667. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Ham, S.; Lee, Y.; Young Suh, G.; Lee, Y.-S. TTC3 contributes to TGF-β1-induced epithelial−mesenchymal transition and myofibroblast differentiation, potentially through SMURF2 ubiquitylation and degradation. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Jung, E.H.; Kim, G.Y.; Kim, B.C.; Lim, J.H.; Woo, C.H. Itch E3 Ubiquitin Ligase Positively Regulates TGF-β Signaling to EMT via Smad7 Ubiquitination. Mol. Cells 2015, 38, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-β (TGF-β) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Soond, S.M.; Chantry, A. Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFΒ signalling and EMT. Oncogene 2011, 30, 2451–2462. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; De Geyter, C.; Jia, Z.; Peng, Y.; Zhang, H. HECTD1 regulates the expression of SNAIL: Implications for epithelial–mesenchymal transition. Int. J. Oncol. 2020, 56, 1186–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhamel, S.; Goyette, M.A.; Thibault, M.P.; Filion, D.; Gaboury, L.; Côté, J.F. The E3 Ubiquitin Ligase HectD1 Suppresses EMT and Metastasis by Targeting the +TIP ACF7 for Degradation. Cell Rep. 2018, 22, 1016–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.; Li, Y.; Wang, M.; Li, X.; Qin, S.; Sun, X.; Liang, R.; Zhang, B.; Du, N.; Xu, C.; et al. FBXW7 suppresses epithelial-mesenchymal transition and chemo-resistance of non-small-cell lung cancer cells by targeting snai1 for ubiquitin-dependent degradation. Cell Prolif. 2018, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y.; et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 2019, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, A.; Onoyama, I.; Sunabori, T.; Kageyama, R.; Okano, H.; Nakayama, K.I. Fbxw7-dependent degradation of notch is required for control of ‘Stemness’ and neuronal-glial differentiation in neural stem cells. J. Biol. Chem. 2011, 286, 13754–13764. [Google Scholar] [CrossRef] [Green Version]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Shen, M.; Zha, Y.L.; Li, W.; Wei, Y.; Blanco, M.A.; Ren, G.; Zhou, T.; Storz, P.; Wang, H.Y.; et al. PKD1 Phosphorylation-Dependent Degradation of SNAIL by SCF-FBXO11 Regulates Epithelial-Mesenchymal Transition and Metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, Y.M.; Yang, C.H.; Cho, S.K.; Lee, J.W.; Cho, M. Functional regulation of Slug/Snail2 is dependent on GSK-3β-mediated phosphorylation. FEBS J. 2012, 279, 2929–2939. [Google Scholar] [CrossRef]
- Zhong, J.; Ogura, K.; Wang, Z.; Inuzuka, H. Degradation of the transcription factor twist, an oncoprotein that promotes cancer metastasis. Discov. Med. 2013, 15, 7–15. [Google Scholar]
- Heo, J.; Eki, R.; Abbas, T. Deregulation of F-box proteins and its consequence on cancer development, progression and metastasis. Semin. Cancer Biol. 2016, 36, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, X.; Zhao, Y.; Jin, Y.; Zheng, J. G-protein-coupled estrogen receptor suppresses the migration of osteosarcoma cells via post-translational regulation of Snail. J. Cancer Res. Clin. Oncol. 2019, 145, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Viñas-Castells, R.; Frías, Á.; Robles-Lanuza, E.; Zhang, K.; Longmore, G.D.; García De Herreros, A.; Díaz, V.M. Nuclear ubiquitination by FBXL5 modulates Snail1 DNA binding and stability. Nucleic Acids Res. 2014, 42, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ding, H.; Cao, J.; Zhang, W. FBXL5 Inhibits Metastasis of Gastric Cancer Through Suppressing Snail1. Cell. Physiol. Biochem. 2015, 35, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Lander, R.; Nordin, K.; LaBonne, C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J. Cell Biol. 2011, 194, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-H.; Su, Y.-H.; Su, W.-H.; Wang, C.-C.; Arbiser, J.; Yang, M.-H. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.H.; Kim, H.; Lee, M.; Yi, J.M.; Kim, R.K.; Uddin, N.; Yoo, K.C.; Kang, J.H.; Choi, M.Y.; Cha, H.J.; et al. FBXL14 abolishes breast cancer progression by targeting CDCP1 for proteasomal degradation. Oncogene 2018, 37, 5794–5809. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; GarcíadeVinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Shenoy, A.K.; Doernberg, S.; Chen, H.; Luo, H.; Shen, H.; Lin, T.; Tarrash, M.; Cai, Q.; Hu, X.; et al. FBXO11 promotes ubiquitination of the Snail family of transcription factors in cancer progression and epidermal development. Cancer Lett. 2015, 362, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Islam, S.; Manne, R.K.; Dinesh, U.S.; Malonia, S.K.; Maity, B.; Boppana, R.; Rapole, S.; Shetty, P.K.; Santra, M.K. F-box protein FBXO16 functions as a tumor suppressor by attenuating nuclear β-catenin function. J. Pathol. 2019, 248, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Ma, C.; Yang, F.; Xu, X.; Jia, J.; Liu, Z. FBXO31 Suppresses Gastric Cancer EMT by Targeting Snail1 for Proteasomal Degradation. Mol. Cancer Res. 2018, 16, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Zhu, C.; Zhao, X.; Chen, C.; Zhang, H.; Yuan, H.; Deng, R.; Dou, J.; Wang, Y.; Huang, J.; et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 2015, 6, 979–994. [Google Scholar] [CrossRef]
- Lin, T.Y.; Hsu, H.Y. Ling Zhi-8 reduces lung cancer mobility and metastasis through disruption of focal adhesion and induction of MDM2-mediated Slug degradation. Cancer Lett. 2016, 375, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ma, X.; Qiu, Y.; Zhu, L.; Lin, Y.; You, Y.; Ma, D.; Qin, Z.; Sun, C.; Zhao, Y.; et al. TRIM50 suppressed hepatocarcinoma progression through directly targeting SNAIL for ubiquitous degradation article. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Chen, N.; Balasenthil, S.; Reuther, J.; Frayna, A.; Wang, Y.; Chandler, D.S.; Abruzzo, L.V.; Rashid, A.; Rodriguez, J.; Lozano, G.; et al. Dear1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-β-driven epithelial-mesenchymal transition. Cancer Discov. 2013, 3, 1172–1189. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Wu, G.; Zhou, Z.; Zhang, X.; Wang, Y.; Song, G.; Ding, E.; Sun, X.; Zhong, L.; Li, S.; et al. RBBP6, a RING finger-domain E3 ubiquitin ligase, induces epithelial–mesenchymal transition and promotes metastasis of colorectal cancer. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Wang, M.; Li, S.; Li, X.; Bai, X.Y.; Xu, Z.; Yang, Y.; Li, B.; Li, Y.; Wu, H. U-box ubiquitin ligase PPIL2 suppresses breast cancer invasion and metastasis by altering cell morphology and promoting SNAI1 ubiquitination and degradation article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; McCollum, C.W.; Bondesson, M.; Quinlan, P.; Thompson, A.; Krishnamurthy, S.; et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xu, L.; Li, C.; Zhao, L.; Ma, Y.; Zheng, H.; Li, Z.; Zhang, Y.; Wang, R.; Liu, Y.; et al. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol. Cancer 2014, 13, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Zhang, Y.; Qu, X.; Che, X.; Guo, T.; Cai, Y.; Li, A.; Li, D.; Li, C.; Wen, T.; et al. E3 Ubiquitin Ligase Cbl-b Prevents Tumor Metastasis by Maintaining the Epithelial Phenotype in Multiple Drug-Resistant Gastric and Breast Cancer Cells. Neoplasia 2017, 19, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.M.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Casas-Pais, A.; Diáz-Diáz, A.; Iglesias, P.; Santamarina, I.; Grana, B.; Calvo, L.; Valladares-Ayerbes, M.; et al. Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P.; Zhang, M.; He, S.; Lu, K.; Chen, Y.; Xing, G.; Lu, Y.; Liu, P.; Li, Y.; Wang, S.; et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 2014, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Sun, C.; Tao, Z.; Yan, S. SMURF1 promotes the proliferation, migration and invasion of gastric cancer cells. Oncol. Rep. 2017, 38, 1806–1814. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, M.; Fukai, Y.; Masuda, N.; Miyazaki, T.; Nakajima, M.; Sohda, M.; Manda, R.; Tsukada, K.; Kato, H.; Kuwano, H. High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res. 2002, 62, 7162–7165. [Google Scholar]
- Salah, Z.; Itzhaki, E.; Aqeilan, R.I. The ubiquitin E3 ligase ITCH enhances breast tumor progression by inhibiting the Hippo tumor suppressor pathway. Oncotarget 2014, 5, 10886–10900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.-F.; Zhang, Q.-G. Inhibition of ITCH Suppresses Proliferation and Induces Apoptosis of Lung Cancer Cells. Cell. Physiol. Biochem. 2018, 48, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Tsuda, H.; Hotta, A.; Kozaki, K.I.; Yoshida, A.; Noh, J.Y.; Ito, K.; Imoto, I.; Inazawa, J. ITCH is a putative target for a novel 20q11.22 amplification detected in anaplastic thyroid carcinoma cells by array-based comparative genomic hybridization. Cancer Sci. 2008, 99, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, X.; Guo, P.; Dong, X.Y.; Sethi, P.; Zhou, W.; Zhou, Z.; Petros, J.; Frierson, H.F.; Vessella, R.L.; et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007, 26, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Z.; Ross, J.S.; Zhou, W.; Dong, J.T. The amplified WWP1 gene is a potential molecular target in breast cancer. Int. J. Cancer 2007, 121, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Akhoondi, S.; Sun, D.; Von Der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dofou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Lu, X.; Liu, Z.; Chen, L.; Xu, Y.; Wang, Y.; Wei, G.; Chen, Y. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015, 6, 6310–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.; Zhang, B.; Yamashita, K.; Bilim, V.; Mai, M.; Fuchs, S.Y.; Minamoto, T. Associations among β-TrCP, an E3 ubiquitin ligase receptor, β-catenin, and NF-κB colorectal cancer. J. Natl. Cancer Inst. 2004, 96, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, S.; Cermak, L.; Pagan, J.K.; Rossi, M.; Martinengo, C.; Di Celle, P.F.; Chapuy, B.; Shipp, M.; Chiarle, R.; Pagano, M. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature 2012, 481, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Pfeffer, S.R.; Sims, M.; Yue, J.; Wang, Y.; Linga, V.G.; Paulus, E.; Davidoff, A.M.; Pfeffer, L.M. The oncogenic microRNA-21 inhibits the tumor suppressive activity of FBXO11 to promote tumorigenesis. J. Biol. Chem. 2015, 290, 6037–6046. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Muzumdar, D.; Shiras, A. Attenuation of Tumor Suppressive Function of FBXO16 Ubiquitin Ligase Activates Wnt Signaling In Glioblastoma. Neoplasia 2019, 21, 106–116. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, Y.; Xu, X.; Xing, H.; Zhang, Y.; Han, F.; Li, W.; Yang, Q.; Zeng, J.; Jia, J.; et al. F-box protein FBXO31 is down-regulated in gastric cancer and negatively regulated by miR-17 and miR-20a. Oncotarget 2014, 5, 6178–6190. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Neilsen, P.M.; Crawford, J.; McKirdy, R.; Lee, J.; Powell, J.A.; Saif, Z.; Martin, J.M.; Lombaerts, M.; Cornelisse, C.J.; et al. FBXO31 is the chromosome 16q24.3 senescence gene, a candidate breast tumor suppressor, and a component of an SCF complex. Cancer Res. 2005, 65, 11304–11313. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-L.; Zheng, W.; Zhao, R.; Zhang, B.; Ma, W. FBXO31 is down-regulated and may function as a tumor suppressor in hepatocellular carcinoma. Oncol. Rep. 2010, 24, 715–720. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Qu, X.; Liu, S.; Yang, X.; Bie, F.; Wang, Y.; Huang, C.; Du, J. Identification of aberrantly expressed F-box proteins in squamous-cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 1509–1521. [Google Scholar] [CrossRef]
- Kogure, N.; Yokobori, T.; Ogata, K.; Altan, B.; Mochiki, E.; Ohno, T.; Toyomasu, Y.; Yanai, M.; Kimura, A.; Yanoma, T.; et al. Low expression of FBXO45 is associated with gastric cancer progression and poor prognosis. Anticancer Res. 2017, 37, 191–196. [Google Scholar] [CrossRef]
- Bueso-Ramos, C.E.; Manshouri, T.; Haidar, M.A.; Huh, Y.O.; Keating, M.J.; Albitar, M. Multiple patterns of MDM-2 deregulation in human leukemias: Implications in leukemogenesis and prognosis. Leuk. Lymphoma 1995, 17, 13–18. [Google Scholar] [CrossRef]
- Tang, Y.; Xuan, Y.; Qiao, G.; Ou, Z.; He, Z.; Zhu, Q.; Liao, M.; Yin, G. MDM2 promotes epithelial–mesenchymal transition through activation of Smad2/3 signaling pathway in lung adenocarcinoma. Onco. Targets. Ther. 2019, 12, 2247–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, D.D.; Wu, Y.P.; Su, D.; Zhou, T.Y.; Gai, R.H.; Fu, Y.Y.; Zheng, L.; He, Q.J.; Zhu, H.; et al. MDM2 promotes epithelial-mesenchymal transition and metastasis of ovarian cancer SKOV3 cells. Br. J. Cancer 2017, 117, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Liu, P.; Ma, X.; Ma, X.; Zhu, L.; Lin, Y.; You, Y.; Yu, W.; Ma, D.; Sun, C.; et al. TRIM50 acts as a novel Src suppressor and inhibits ovarian cancer progression. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Lott, S.T.; Chen, N.; Chandler, D.S.; Yang, Q.; Wang, L.; Rodriguez, M.; Xie, H.; Balasenthil, S.; Buchholz, T.A.; Sahin, A.A.; et al. DEAR1 is a dominant regulator of acinar morphogenesis and an independent predictor of local recurrence-free survival in early-onset breast cancer. PLoS Med. 2009, 6, e1000068. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Post, S.M.; Solis, L.M.; Xiong, S.; Yang, P.; Chen, N.; Wistuba, I.I.; Killary, A.M.; Lozano, G. Loss of the novel tumour suppressor and polarity gene Trim62 (Dear1) synergizes with oncogenic Ras in invasive lung cancer. J. Pathol. 2014, 234, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshitake, Y.; Nakatsura, T.; Monji, M.; Senju, S.; Matsuyoshi, H.; Tsukamoto, H.; Hosaka, S.; Komori, H.; Fukuma, D.; Ikuta, Y.; et al. Proliferation potential-related protein, an ideal esophageal cancer antigen for immunotherapy, identified using complementary DNA microarray analysis. Clin. Cancer Res. 2004, 10, 6437–6448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motadi, L.R.; Moela, P.; Choene, M.S. Silencing RBBP6 (retinoblastoma binding protein 6) sensitizes breast cancer cells to staurosporine and camptothecin-induced cell death. Cancer Res. 2014, 74, 344. [Google Scholar] [CrossRef]
- Motadi, L.R.; Bhoola, K.D.; Dlamini, Z. Expression and function of retinoblastoma binding protein 6 (RBBP6) in human lung cancer. Immunobiology 2011, 216, 1065–1073. [Google Scholar] [CrossRef]
- Moela, P.; Motadi, L.R. RBBP6: A potential biomarker of apoptosis induction in human cervical cancer cell lines. Onco. Targets. Ther. 2016, 9, 4721–4735. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.T.; Lin, Y.C.; Lin, P.H.; Shih, J.Y.; Chou, C.W.; Huang, W.J.; Yang, Y.C.; Hsiao, P.W.; Chen, C.C. Induction of c-Cbl contributes to anti-cancer effects of HDAC inhibitor in lung cancer. Oncotarget 2015, 6, 12481–12492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Xu, L.; Che, X.; Li, C.; Xu, L.; Hou, K.; Fan, Y.; Wen, T.; Qu, X.; Liu, Y. E3 ubiquitin ligases Cbl-b and c-Cbl downregulate PD-L1 in EGFR wild-type non-small cell lung cancer. FEBS Lett. 2018, 592, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zhang, S.; Wudu, M.; Ren, H.; Xu, Y.; Zhang, Q.; Qiu, X. CBLL1 is highly expressed in non-small cell lung cancer and promotes cell proliferation and invasion. Thorac. Cancer 2019, 10, 1759–7714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, D.C.; Malik, B.; Bao, H.F.; Yu, L.; Jain, L. Regulation of epithelial sodium channel trafficking by ubiquitination. Proc. Am. Thorac. Soc. 2010, 7, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, A.; Marchese, A.; Henry, P.; Rotin, D.; Morrione, A. The Grb10/Nedd4 Complex Regulates Ligand-Induced Ubiquitination and Stability of the Insulin-Like Growth Factor I Receptor. Mol. Cell. Biol. 2003, 23, 3363–3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandhoke, A.S.; Karve, K.; Dadakhujaev, S.; Netherton, S.; Deng, L.; Bonni, S. The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial-mesenchymal transition in a sumoylation-regulated manner. Cell Death Differ. 2016, 23, 876–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Shi, L.; Yang, C.; Ge, Y.; Lin, L.; Fan, H.; He, Y.; Zhang, D.; Miao, Y.; Yang, L. miR-1254 inhibits cell proliferation, migration, and invasion by down-regulating Smurf1 in gastric cancer. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-β signaling by modulating Smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef]
- Wu, X.; Shen, Q.T.; Oristian, D.S.; Lu, C.P.; Zheng, Q.; Wang, H.W.; Fuchs, E. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 2011, 144, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Díaz, V.M.; de Herreros, A.G. F-box proteins: Keeping the epithelial-to-mesenchymal transition (EMT) in check. Semin. Cancer Biol. 2016, 36, 71–79. [Google Scholar] [CrossRef]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Wartz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jung, S.M.; Yang, K.M.; Bae, E.; Ahn, S.G.; Park, J.S.; Seo, D.; Kim, M.; Ha, J.; Lee, J.; et al. A20 promotes metastasis of aggressive basal-like breast cancers through multi-monoubiquitylation of Snail1. Nat. Cell Biol. 2017, 19, 1260–1273. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitmann, J.S.; Hagelstein, I.; Hinterleitner, C.; Roerden, M.; Jung, G.; Salih, H.R.; Märklin, M.; Kauer, J. Identification of CD318 (CDCP1) as novel prognostic marker in AML. Ann. Hematol. 2020, 99, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Saiga, T.; Fukuda, T.; Matsumoto, M.; Tada, H.; Okano, H.J.; Okano, H.; Nakayama, K.I. Fbxo45 Forms a Novel Ubiquitin Ligase Complex and Is Required for Neuronal Development. Mol. Cell. Biol. 2009, 29, 3529–3543. [Google Scholar] [CrossRef] [Green Version]
- Chène, P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef]
- Hauck, P.M.; Wolf, E.R.; Olivos, D.J.; Batuello, C.N.; McElyea, K.C.; McAtarsney, C.P.; Cournoyer, R.M.; Sandusky, G.E.; Mayo, L.D. Early-stage metastasis requires Mdm2 and not p53 gain of function. Mol. Cancer Res. 2017, 15, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Yan, C.; Huang, Y.; Shi, D.; Fu, Z.; Qiu, J.; Yin, Y. Mouse double minute 2 (MDM2) upregulates Snail expression and induces epithelial-to-mesenchymal transition in breast cancer cells in vitro and in vivo. Oncotarget 2016, 7, 37177–37191. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Chen, N.; Balasenthil, S.; Reuther, J.; Killary, A.M.N. DEAR1, a novel tumor suppressor that regulates cell polarity and epithelial plasticity. Cancer Res. 2014, 74, 5683–5689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Rao, J.; Lou, X.; Zhai, J.; Ni, Z.; Wang, X. Upregulated TRIM11 Exerts its Oncogenic Effects in Hepatocellular Carcinoma Through Inhibition of P53. Cell. Physiol. Biochem. 2017, 44, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Xu, C.; Zhang, X.; Huang, L.; Zheng, C.; Chen, H.; Wang, Y.; Ju, H.; Yao, Q. TRIM11 upregulation contributes to proliferation, invasion, and EMT of hepatocellular carcinoma cells. Oncol. Res. 2017, 25, 691–699. [Google Scholar] [CrossRef]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Sung, Y.J.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Thien, C.B.F.; Langdon, W.Y. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef]
- Liyasova, M.S.; Ke, M.; Lipkowitz, S. Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity—Opportunities for cancer treatment. Clin. Cancer Res. 2015, 21, 1789–1794. [Google Scholar] [CrossRef] [Green Version]
- Davies, G.C.; Ryan, P.E.; Rahman, L.; Zajac-Kaye, M.; Lipkowitz, S. EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins. Oncogene 2006, 25, 6497–6509. [Google Scholar] [CrossRef] [Green Version]
- Daniels, S.R.; Liyasova, M.; Kales, S.C.; Nau, M.M.; Ryan, P.E.; Green, J.E.; Lipkowitz, S. Loss of function Cbl-c mutations in solid tumors. PLoS ONE 2019, 14, e0219143. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, M.; Lin, R.; Zhan, M.; Xu, S.; Huang, X.; Xu, C.; Chen, W.; Yao, Y.; Mohan, M.; et al. PLEK2 promotes gallbladder cancer invasion and metastasis through EGFR/CCL2 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, M.; Jing-Song, F.; Ramachandran, S.; Guy, G.R.; Sivaraman, J. Dimeric switch of Hakai-truncated monomers during substrate recognition: Insights from solution studies and nmr structure. J. Biol. Chem. 2014, 289, 25611–25623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, F.; Tushir, J.S.; Fujita, Y.; D’Souza-Schorey, C. Lysosomal Targeting of E-Cadherin: A Unique Mechanism for the Down-Regulation of Cell-Cell Adhesion during Epithelial to Mesenchymal Transitions. Mol. Cell. Biol. 2005, 25, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Díaz, A.; Casas-Pais, A.; Calamia, V.; Castosa, R.; Martinez-Iglesias, O.; Roca-Lema, D.; Santamarina, I.; Valladares-Ayerbes, M.; Calvo, L.; Chantada, V.; et al. Proteomic Analysis of the E3 Ubiquitin-Ligase Hakai Highlights a Role in Plasticity of the Cytoskeleton Dynamics and in the Proteasome System. J. Proteome Res. 2017, 16, 2773–2788. [Google Scholar] [CrossRef]
- Díaz-Díaz, A.; Roca-Lema, D.; Casas-Pais, A.; Romay, G.; Colombo, G.; Concha, Á.; Graña, B.; Figueroa, A. Heat shock protein 90 chaperone regulates the E3 ubiquitin-ligase hakai protein stability. Cancers 2020, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, A.; Kotani, H.; Toda, Y.; Mazan-Mamczarz, K.; Mueller, E.C.; Otto, A.; Disch, L.; Norman, M.; Ramdasi, R.M.; Keshtgar, M.; et al. Novel roles of Hakai in cell proliferation and oncogenesis. Mol. Biol. Cell 2009, 20, 3533–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, A.; Fujita, Y.; Gorospe, M. Hacking RNA: Hakai promotes tumorigenesis by enhancing the RNA-binding function of PSF. Cell Cycle 2009, 8, 3648–3651. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rigueiro, T.; Valladares-Ayerbes, M.; Haz-Conde, M.; Blanco, M.; Aparicio, G.; Fernández-Puente, P.; Blanco, F.J.; Lorenzo, M.J.; Aparicio, L.A.; Figueroa, A. A novel procedure for protein extraction from formalin-fixed paraffin-embedded tissues. Proteomics 2011, 11, 2555–2559. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, L.A.; Castosa, R.; Haz-Conde, M.; Rodríguez, M.; Blanco, M.; Valladares, M.; Figueroa, A. Role of the microtubule-targeting drug vinflunine on cell-cell adhesions in bladder epithelial tumour cells. BMC Cancer 2014, 14, 1–13. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein | EMT-Related Substrates | Functional Roles | Reference |
|---|---|---|---|
| Smurf 1 | Smad1, Smad2, DAB21P, ARHGAP26 | Promotes cell migration and invasion and metastasis | [63,64] |
| Smurf 2 | Smad1, Smad2, Smurf1, TβRI, | Up-regulation of N-cadherin. Knockdown of Smurf2 enhances Smurf1 levels, increasing migration and metastasis in vivo | [65,66,67] |
| Ttc3 | Smurf2 | Implicated in TGF-β1-induced EMT | [67] |
| Itch | Smad7 | Positively regulates EMT induced by TGF-β | [68] |
| WWP1 | TGF- β type I Receptor | Negatively regulates TGF-β tumor suppressor pathway | [69] |
| WWP2 | Smads | TGFβ-dependent transcription and EMT | [70] |
| HectD1 | ACF7, Snail | Inhibits the reorganization of microtubule and promote epithelial phenotypes | [71,72] |
| Fbxw7 | Zeb1, Snai1, c-myc, Notch | Tumor suppressor. Suppression of EMT and chemoresistance | [73,74,75,76] |
| Fbxw1 (β-TrCP1) | Snai1, Snail2 | Oncogenic or tumor-suppressor functions in a tissue-specific manner | [77,78,79,80] |
| Fbxl5 | Snai1 | Inhibition of gastric cancer metastasis | [81,82,83] |
| Fbxl14 | Snai1, Snail2, Zeb2, Twist, CDCP1 | Suppression of EMT, stemness and metastasis | [84,85,86] |
| Fbxo11 | Snai1, Snail2, Scartch | Blocks EMT in breast cancer. Down-regulated by TGF-β | [87,88] |
| Fbxo16 | β-catenin | Inhibits EMT and prevent malignancy | [89] |
| Fbxo31 | Snai1 | Inhibition of gastric cancer metastasis | [90] |
| Fbxo45 | Snai1, Snail2, Zeb2, Twist | Suppresses EMT characteristics. Negative regulated by miR-27a* | [91] |
| Mdm2 | Snail2 | rLZ-8,a recombinant protein of medicinal mushroom G. lucidum, promoted the degradation of Snail1 by MDM, preventing lung cancer cell proliferation in vivo | [92] |
| Trim50 | Snail1 | Antitumor and anti-EMT effect in hepatocarcinoma | [93] |
| Trim62 | SMAD3 | Depletion of EMT TFs | [94] |
| Rbbp6 | IκBα | Activation of NF-κB-and EMT | [95] |
| Ppil2 | Snai1 | Repression of EMT in breast cancer | [96] |
| Cbl | EGFR | Inhibition of EMT, invasion and migration by the degradation of EGFR in response to ERβ1 | [97] |
| Cbl-b | EGFR, IGF-IR | Maintenance of the epithelial phenotype by inhibition of cell migration in gastric and breast multidrug resistant cancer cells | [98,99] |
| Hakai | E-cadherin | Disruption of cell–cell contacts and induction of EMT in vitro and in vivo | [58,100,101] |
| Protein | Associated Cancers | Reference |
|---|---|---|
| Smurf 1 | Overexpressed in colorectal and gastric cancer | [102,103] |
| Smurf 2 | Overexpressed in esophageal squamous cell carcinoma | [104] |
| Itch | Overexpressed in breast cancer, lung cancer and amplification in 20q11.22 in thyroid carcinoma | [105,106,107] |
| WWP1 | Overexpressed in prostate cancer and breast cancer | [108,109] |
| HectD1 | Downregulated in breast cancer and cervical cancer | [71,72] |
| Fbxw7 | Inactivated by mutation in bile duct, blood, endometrium, colon and stomach cancer. | [110] |
| Downregulated in cholangiocarcinoma | [111] | |
| Fbxw1 (β-TrCP1) | Expression increased in colorectal cancer | [112] |
| Fbxl5 | Downregulated in gastric cancer | [83] |
| Fbxl14 | Downregulated in breast cancer | [86] |
| Fbxo11 | Deleted or mutated in diffuse large B-cell lymphoma | [113] |
| Expression decreased in skin cancer, glioblastoma and prostate cancer | [114] | |
| Reduced expression is correlated with adverse clinical outcome in lung cancer | [88] | |
| Fbxo16 | Expression attenuated with cancer progression in breast cancer | [89] |
| Downregulated in glioblastoma | [115] | |
| Fbxo31 | Downregulated in gastric cancer, breast cancer and hepatocellular | [116,117,118] |
| Fbxo45 | Overexpressed in squamous-cell lung carcinoma (SCLC) | [119] |
| Downregulated in gastric cancer | [120] | |
| Mdm2 | Overexpressed in leukemias and on-third of sarcomas, lung cancer and ovarian cancer | [121,122,123] |
| Trim50 | Downregulated in hepatocarcinoma and ovarian cancer | [93,124] |
| Trim62 | Mutated and downregulated in breast cancer | [125] |
| Downregulated in non-small cell lung cancer (NSCLC) | [126] | |
| Rbbp6 | Overexpressed in esophageal cancer, breast cancer, lung cancer, cervical cancer and colorectal cancer | [95,127,128,129,130] |
| Ppil2 | Downregulated in breast cancer | [96] |
| Cbl | Downregulated in lung cancer | [131] |
| Cbl-b | Downregulated in gastric and breast cancer | [98,99] |
| Associated with better outcome in NSCLC patients | [132] | |
| Hakai | Overexpressed in colorectal cancer and NSCLC | [101,133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Alonso, A.; Casas-Pais, A.; Roca-Lema, D.; Graña, B.; Romay, G.; Figueroa, A. Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer. Cancers 2020, 12, 3093. https://doi.org/10.3390/cancers12113093
Rodríguez-Alonso A, Casas-Pais A, Roca-Lema D, Graña B, Romay G, Figueroa A. Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer. Cancers. 2020; 12(11):3093. https://doi.org/10.3390/cancers12113093
Chicago/Turabian StyleRodríguez-Alonso, Andrea, Alba Casas-Pais, Daniel Roca-Lema, Begoña Graña, Gabriela Romay, and Angélica Figueroa. 2020. "Regulation of Epithelial–Mesenchymal Plasticity by the E3 Ubiquitin-Ligases in Cancer" Cancers 12, no. 11: 3093. https://doi.org/10.3390/cancers12113093