YC-1 Antagonizes Wnt/β-Catenin Signaling Through the EBP1 p42 Isoform in Hepatocellular Carcinoma


Abstract
:1. Introduction
2. Results
2.1. YC-1 Inhibits Wnt Signaling and Suppresses Cell Proliferation in HCC
2.2. YC-1 Enhances the Recruitment of EBP1 to Interact with the β-Catenin/TCF4 Complex
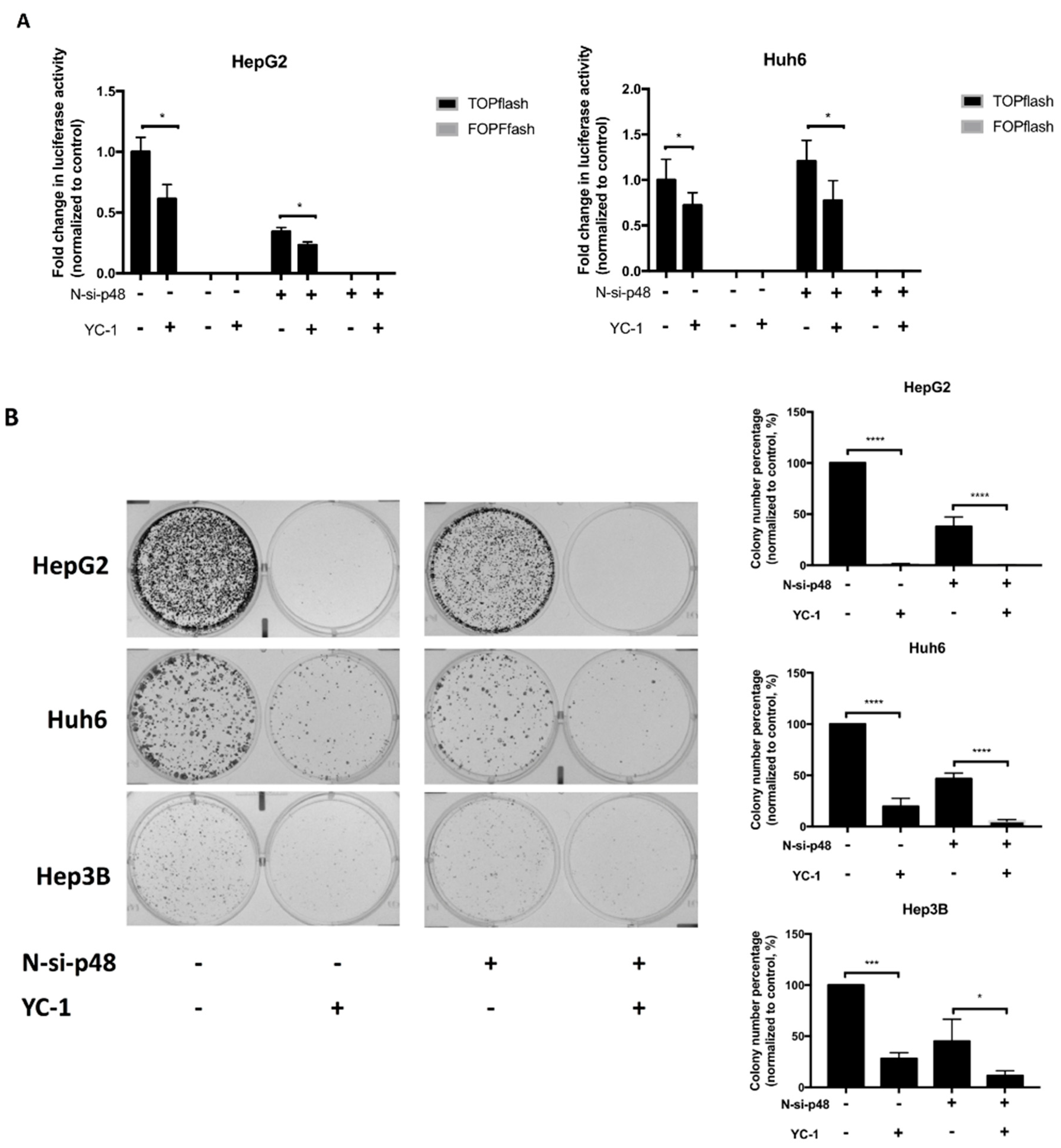
2.3. Knockdown of EBP1 Inhibits the Suppressive Effect of YC-1 in HCC
2.4. YC-1 Promotes the Interaction of EBP1 p42 with the β-Catenin/TCF4 Complex
2.5. YC-1 Inhibits Colony Formation through EBP1 p42-Mediated Suppression of the Wnt/β-Catenin Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. The Delivery of Short Hairpin RNA (shRNA) or Dicer-Substrate siRNA (DsiRNA)
4.3. TCF/LEF Luciferase Assay
4.4. Cell Viability Assay
4.5. Clonogenic Assay
4.6. Immunocytochemical (ICC) Staining
4.7. Nuclear Extraction
4.8. Coimmunoprecipitation Assays and Mass Spectrometry
4.9. Western Blotting
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| YC-1 | 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole |
| TCF4 | T cell-specific factor 4 |
| EBP1 | ErbB3 binding protein 1 |
| HCC | hepatocellular carcinoma |
| STF | Super-TOPflash |
| LOPAC | Library of Pharmacologically Active Compounds |
| co-IP | co-immunoprecipitation |
| HIF-1α | hypoxia-inducible factor 1-α |
| MTS | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
References
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Signaling pathway/molecular targets and new targeted agents under development in hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 6005–6017. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Gammons, M.; Bienz, M. Multiprotein complexes governing wnt signal transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.M.; Yan, M.D.; Shih, Y.L.; Yu, P.N.; Kuo, C.C.; Lin, W.C.; Li, H.J.; Lin, Y.W. Sox1 functions as a tumor suppressor by antagonizing the wnt/beta-catenin signaling pathway in hepatocellular carcinoma. Hepatology 2012, 56, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chua, M.S.; Grepper, S.; So, S. Small molecule antagonists of tcf4/beta-catenin complex inhibit the growth of hcc cells in vitro and in vivo. Int. J. Cancer 2010, 126, 2426–2436. [Google Scholar]
- Lin, H.H.; Feng, W.C.; Lu, L.C.; Shao, Y.Y.; Hsu, C.H.; Cheng, A.L. Inhibition of the wnt/beta-catenin signaling pathway improves the anti-tumor effects of sorafenib against hepatocellular carcinoma. Cancer Lett. 2016, 381, 58–66. [Google Scholar] [CrossRef]
- Eguchi, M.; Nguyen, C.; Lee, S.C.; Kahn, M. ICG-001, a novel small molecule regulator of tcf/beta-catenin transcription. Med. Chem. 2005, 1, 467–472. [Google Scholar] [CrossRef]
- Tsui, L.; Fong, T.H.; Wang, I.J. The effect of 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (yc-1) on cell viability under hypoxia. Mol. Vis. 2013, 19, 2260–2273. [Google Scholar] [PubMed]
- Hsieh, K.Y.; Wei, C.K.; Wu, C.C. Yc-1 prevents tumor-associated tissue factor expression and procoagulant activity in hypoxic conditions by inhibiting p38/nf-kappab signaling pathway. Int. J. Mol. Sci. 2019, 20, 244. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.K.; Yang, Z.F.; Lam, S.P.; Lam, C.T.; Ngai, P.; Tam, K.H.; Poon, R.T.; Fan, S.T. Inhibition of stat3 activity by yc-1 enhances chemo-sensitivity in hepatocellular carcinoma. Cancer Biol. Ther. 2007, 6, 1900–1907. [Google Scholar] [CrossRef]
- Kong, J.; Kong, F.; Gao, J.; Zhang, Q.; Dong, S.; Gu, F.; Ke, S.; Pan, B.; Shen, Q.; Sun, H.; et al. Yc-1 enhances the anti-tumor activity of sorafenib through inhibition of signal transducer and activator of transcription 3 (stat3) in hepatocellular carcinoma. Mol. Cancer 2014, 13, 7. [Google Scholar] [CrossRef]
- Liu, Z.; Ahn, J.Y.; Liu, X.; Ye, K. Ebp1 isoforms distinctively regulate cell survival and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 10917–10922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.R.; Chang, Y.S.; Park, W.S.; Ahn, J.Y. Opposing roles of the two isoforms of erbb3 binding protein 1 in human cancer cells. Int. J. Cancer 2016, 139, 1202–1208. [Google Scholar] [CrossRef]
- Nguyen, D.Q.; Hoang, D.H.; Nguyen Vo, T.T.; Huynh, V.; Ghoda, L.; Marcucci, G.; Nguyen, L.X.T. The role of erbb3 binding protein 1 in cancer: Friend or foe? J. Cell. Physiol. 2018, 233, 9110–9120. [Google Scholar] [CrossRef]
- Zhang, Y.; Woodford, N.; Xia, X.; Hamburger, A.W. Repression of e2f1-mediated transcription by the erbb3 binding protein ebp1 involves histone deacetylases. Nucleic Acids Res. 2003, 31, 2168–2177. [Google Scholar] [CrossRef]
- Chun, Y.S.; Yeo, E.J.; Park, J.W. Versatile pharmacological actions of yc-1: Anti-platelet to anticancer. Cancer Lett. 2004, 207, 1–7. [Google Scholar] [CrossRef]
- Yeo, E.J.; Chun, Y.S.; Cho, Y.S.; Kim, J.; Lee, J.C.; Kim, M.S.; Park, J.W. Yc-1: A potential anticancer drug targeting hypoxia-inducible factor 1. J. Natl. Cancer Inst. 2003, 95, 516–525. [Google Scholar] [CrossRef]
- Wang, S.W.; Pan, S.L.; Guh, J.H.; Chen, H.L.; Huang, D.M.; Chang, Y.L.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. Yc-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] exhibits a novel antiproliferative effect and arrests the cell cycle in g0-g1 in human hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2005, 312, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.; Bush, K.M.; Knoepfler, P.S. Erbb3-binding protein 1 (ebp1) is a novel developmental pluripotency-associated-4 (dppa4) cofactor in human pluripotent cells. Stem Cells 2018, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- He, H.C.; Ling, X.H.; Zhu, J.G.; Fu, X.; Han, Z.D.; Liang, Y.X.; Deng, Y.H.; Lin, Z.Y.; Chen, G.; Chen, Y.F.; et al. Down-regulation of the erbb3 binding protein 1 in human bladder cancer promotes tumor progression and cell proliferation. Mol. Biol. Rep. 2013, 40, 3799–3805. [Google Scholar] [CrossRef]
- Kim, C.K.; Nguyen, T.L.; Joo, K.M.; Nam, D.H.; Park, J.; Lee, K.H.; Cho, S.W.; Ahn, J.Y. Negative regulation of p53 by the long isoform of erbb3 binding protein ebp1 in brain tumors. Cancer Res. 2010, 70, 9730–9741. [Google Scholar] [CrossRef]
- Ko, H.R.; Nguyen, T.L.; Kim, C.K.; Park, Y.; Lee, K.H.; Ahn, J.Y. P42 ebp1 functions as a tumor suppressor in non-small cell lung cancer. BMB Rep. 2015, 48, 159–165. [Google Scholar] [CrossRef]
- Liu, L.; Li, X.D.; Chen, H.Y.; Cui, J.S.; Xu, D.Y. Significance of ebp1 and p53 protein expression in cervical cancer. Genet. Mol. Res. 2015, 14, 11860–11866. [Google Scholar] [CrossRef]
- Nguyen le, X.T.; Zhu, L.; Lee, Y.; Ta, L.; Mitchell, B.S. Expression and role of the erbb3-binding protein 1 in acute myelogenous leukemic cells. Clin. Cancer Res. 2016, 22, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Luo, Y.; Tian, Z.; Gu, L.; Xia, S.C.; Yu, Y. Expression of erbb3 binding protein 1 (ebp1) in salivary adenoid cystic carcinoma and its clinicopathological relevance. BMC Cancer 2012, 12, 499. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Wang, Y.; Zhan, P.; Liu, C.; Mao, J.H.; Wei, G. Distinct interactions of ebp1 isoforms with fbxw7 elicits different functions in cancer. Cancer Res. 2017, 77, 1983–1996. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.W.; Jelovac, D.; Nakanishi, T.; Yu, M.H.; Akinmade, D.; Goloubeva, O.; Ross, D.D.; Brodie, A.; Hamburger, A.W. The erbb3-binding protein ebp1 suppresses androgen receptor-mediated gene transcription and tumorigenesis of prostate cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9890–9895. [Google Scholar] [CrossRef]
- Zhang, Y.; Linn, D.; Liu, Z.; Melamed, J.; Tavora, F.; Young, C.Y.; Burger, A.M.; Hamburger, A.W. Ebp1, an erbb3-binding protein, is decreased in prostate cancer and implicated in hormone resistance. Mol. Cancer Ther. 2008, 7, 3176–3186. [Google Scholar] [CrossRef]
- Hu, B.; Xiong, Y.; Ni, R.; Wei, L.; Jiang, D.; Wang, G.; Wu, D.; Xu, T.; Zhao, F.; Zhu, M.; et al. The downregulation of erbb3 binding protein 1 (ebp1) is associated with poor prognosis and enhanced cell proliferation in hepatocellular carcinoma. Mol. Cell. Biochem. 2014, 396, 175–185. [Google Scholar] [CrossRef]
- Ko, H.R.; Kim, C.K.; Ahn, J.Y. Phosphorylation of the n-terminal domain of p48 ebp1 by cdk2 is required for tumorigenic function of p48. Mol. Carcinog. 2015, 54, 1283–1291. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Liu, X.; Liu, Z.; Pereira, L.; Cheng, D.; Peng, J.; Wade, P.A.; Hamburger, A.W.; Ye, K. Nuclear akt associates with pkc-phosphorylated ebp1, preventing DNA fragmentation by inhibition of caspase-activated dnase. EMBO J. 2006, 25, 2083–2095. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, S.B.; Nguyen, T.L.; Lee, K.H.; Um, S.H.; Kim, J.; Ahn, J.Y. Long isoform of erbb3 binding protein, p48, mediates protein kinase b/akt-dependent hdm2 stabilization and nuclear localization. Exp. Cell Res. 2012, 318, 136–143. [Google Scholar] [CrossRef]
- Tu, K.; Zheng, X.; Zhou, Z.; Li, C.; Zhang, J.; Gao, J.; Yao, Y.; Liu, Q. Recombinant human adenovirus-p53 injection induced apoptosis in hepatocellular carcinoma cell lines mediated by p53-fbxw7 pathway, which controls c-myc and cyclin e. PLoS ONE 2013, 8, e68574. [Google Scholar] [CrossRef]
- Lessor, T.J.; Hamburger, A.W. Regulation of the erbb3 binding protein ebp1 by protein kinase C. Mol. Cell. Endocrinol. 2001, 175, 185–191. [Google Scholar] [CrossRef]
- Akinmade, D.; Lee, M.; Zhang, Y.; Hamburger, A.W. Ebp1-mediated inhibition of cell growth requires serine 363 phosphorylation. Int. J. Oncol. 2007, 31, 851–858. [Google Scholar] [CrossRef]
- Chang, M.S.; Lee, W.S.; Teng, C.M.; Lee, H.M.; Sheu, J.R.; Hsiao, G.; Lin, C.H. Yc-1 increases cyclo-oxygenase-2 expression through protein kinase G- and p44/42 mitogen-activated protein kinase-dependent pathways in a549 cells. Br. J. Pharmacol. 2002, 136, 558–567. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| Scrambled shRNA | CCGGTGTTCGCATTATCCGAACCATCTCGAGATGGTTCGGATAATGCGAACATTTT |
| shEBP1#1 | CCGGCCACCAGCATTTCGGTAAATACTCGAGTATTTACCGAAATGCTGGTGGTTTTTG |
| shEBP1#2 | CCGGCGCTAATGTAGCTCACACTTTCTCGAGAAAGTGTGAGCTACATTAGCGTTTTTG |
| shEBP1#3 | CCGGCCTGGTCGTGACCAAGTATAACTCGAGTTATACTTGGTCACGACCAGGTTTTTG |
| shEBP1#4 | CCGGAGGACAGAGAACCACTATTTACTCGAGTAAATAGTGGTTCTCTGTCCTTTTTTG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.-Y.; Shih, Y.-L.; Lin, S.-P.; Hsieh, T.-Y.; Lin, Y.-W. YC-1 Antagonizes Wnt/β-Catenin Signaling Through the EBP1 p42 Isoform in Hepatocellular Carcinoma. Cancers 2019, 11, 661. https://doi.org/10.3390/cancers11050661
Wu J-Y, Shih Y-L, Lin S-P, Hsieh T-Y, Lin Y-W. YC-1 Antagonizes Wnt/β-Catenin Signaling Through the EBP1 p42 Isoform in Hepatocellular Carcinoma. Cancers. 2019; 11(5):661. https://doi.org/10.3390/cancers11050661
Chicago/Turabian StyleWu, Ju-Yun, Yu-Lueng Shih, Shih-Ping Lin, Tsai-Yuan Hsieh, and Ya-Wen Lin. 2019. "YC-1 Antagonizes Wnt/β-Catenin Signaling Through the EBP1 p42 Isoform in Hepatocellular Carcinoma" Cancers 11, no. 5: 661. https://doi.org/10.3390/cancers11050661