Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model

 ,
, 
Abstract
:1. Introduction
2. Results
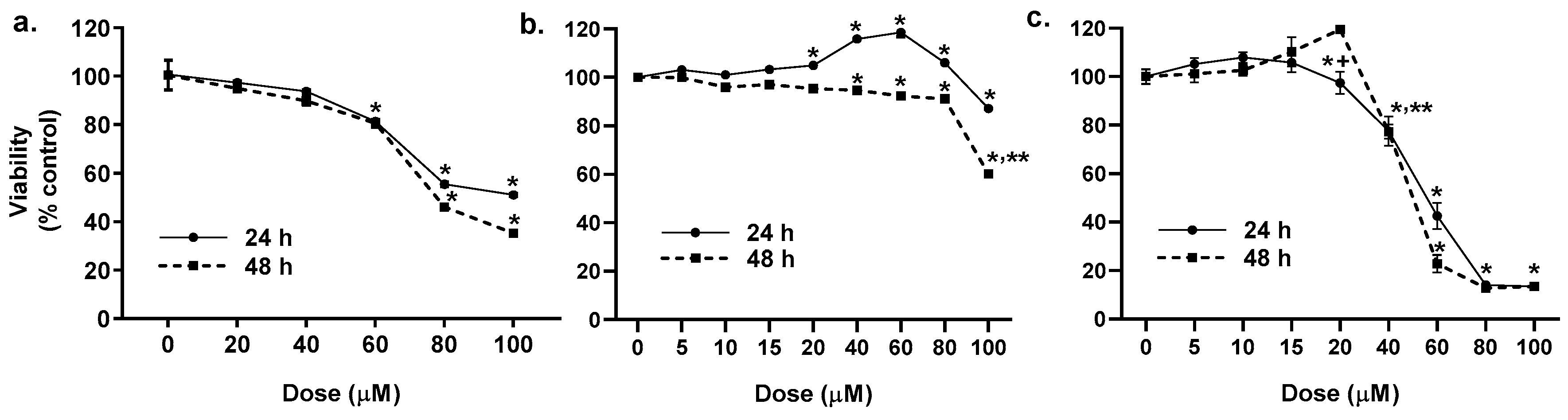
2.1. Cytotoxicity of the LMW PAHs in Lung Epithelial Cells and Macrophages
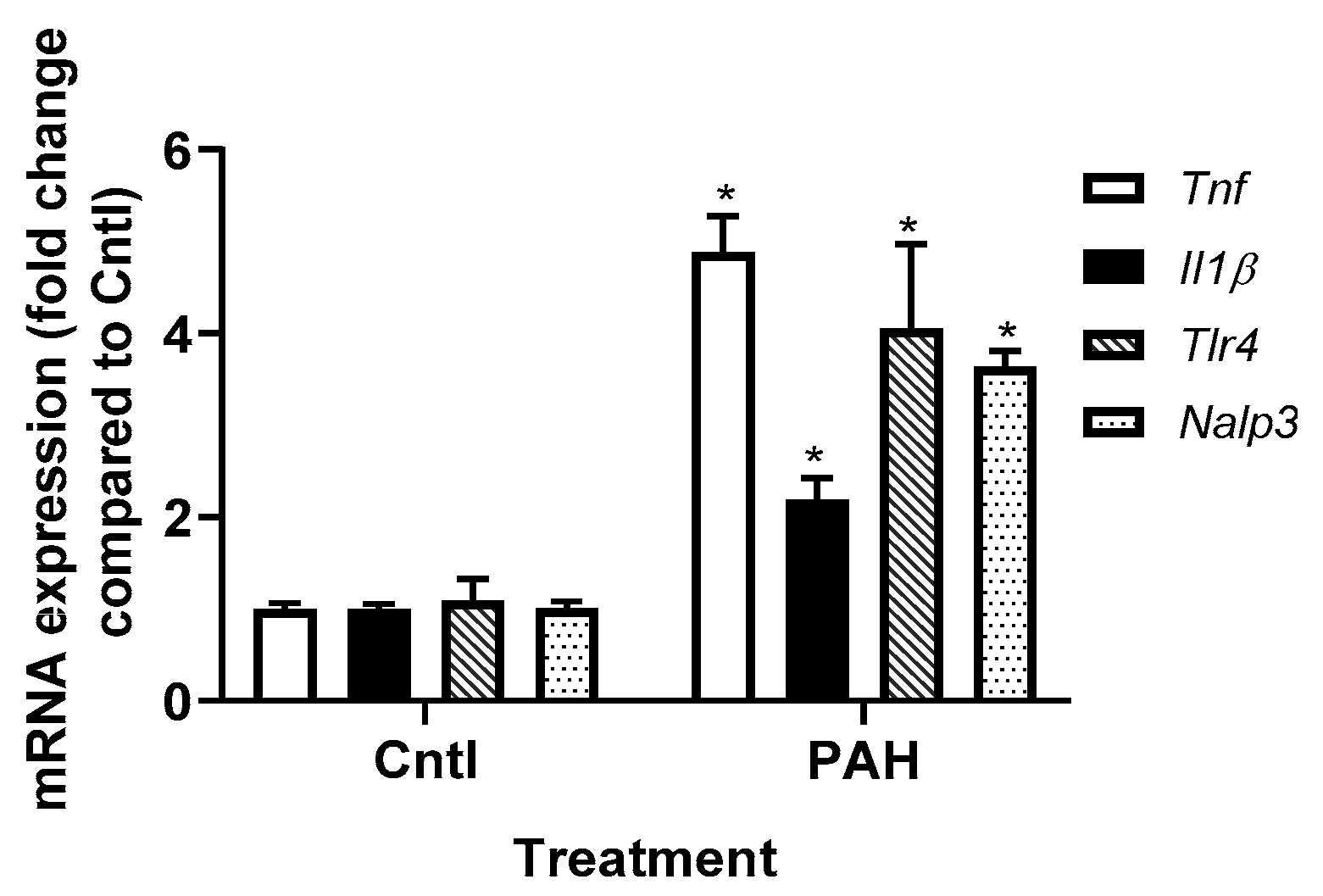
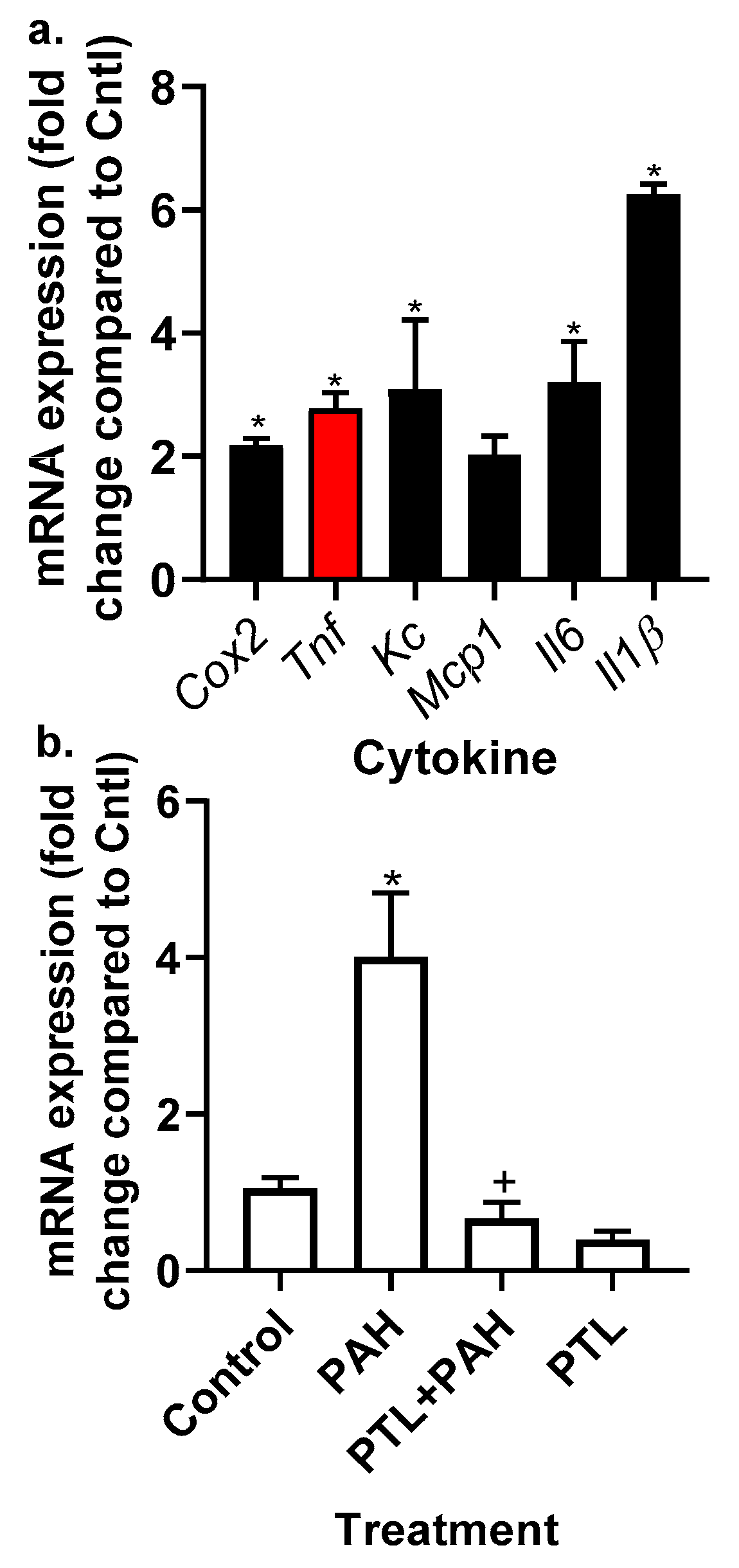
2.2. PAH-Induced Cytokine and Chemokine mRNA Expression in C10 Cells
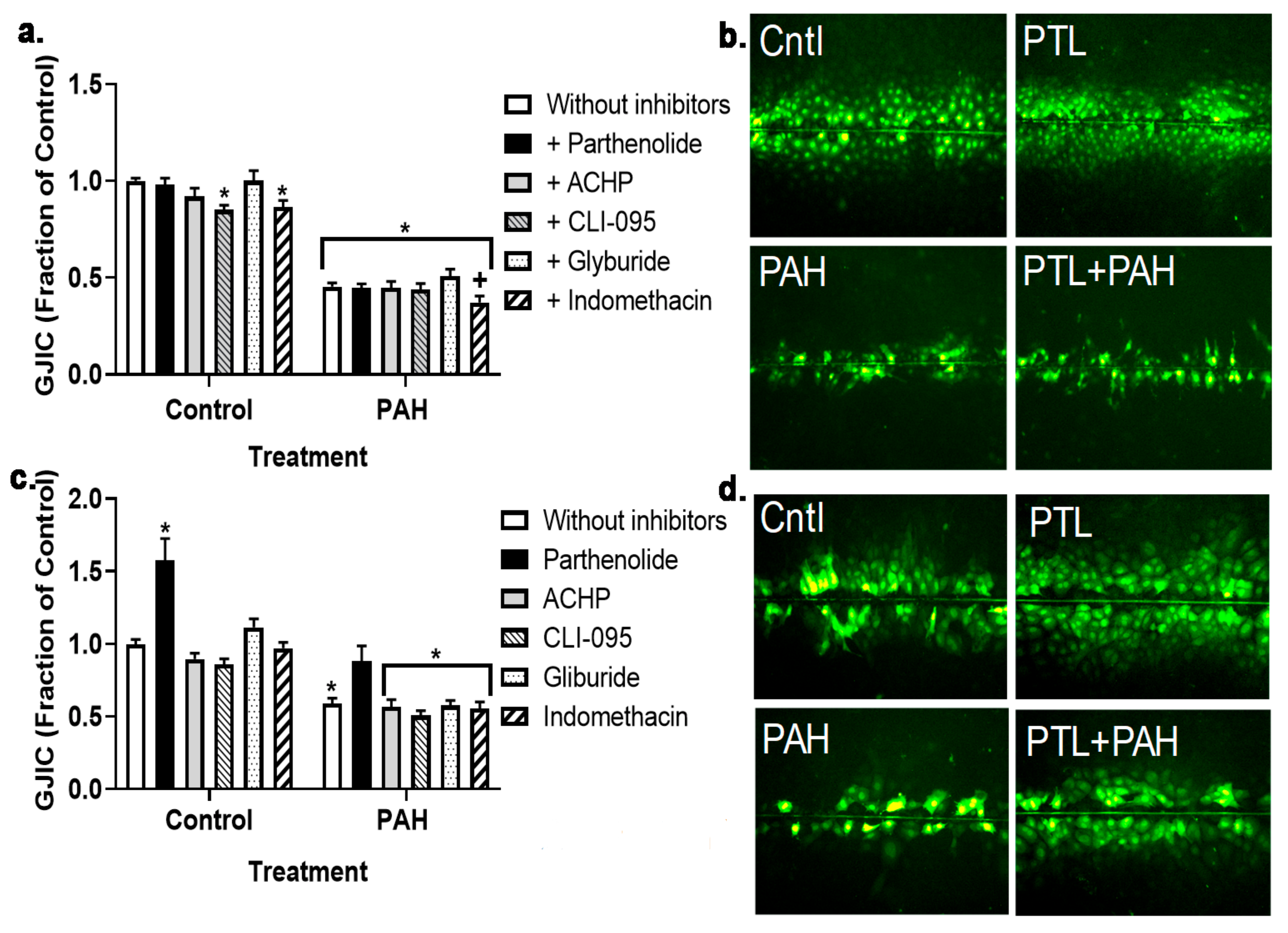
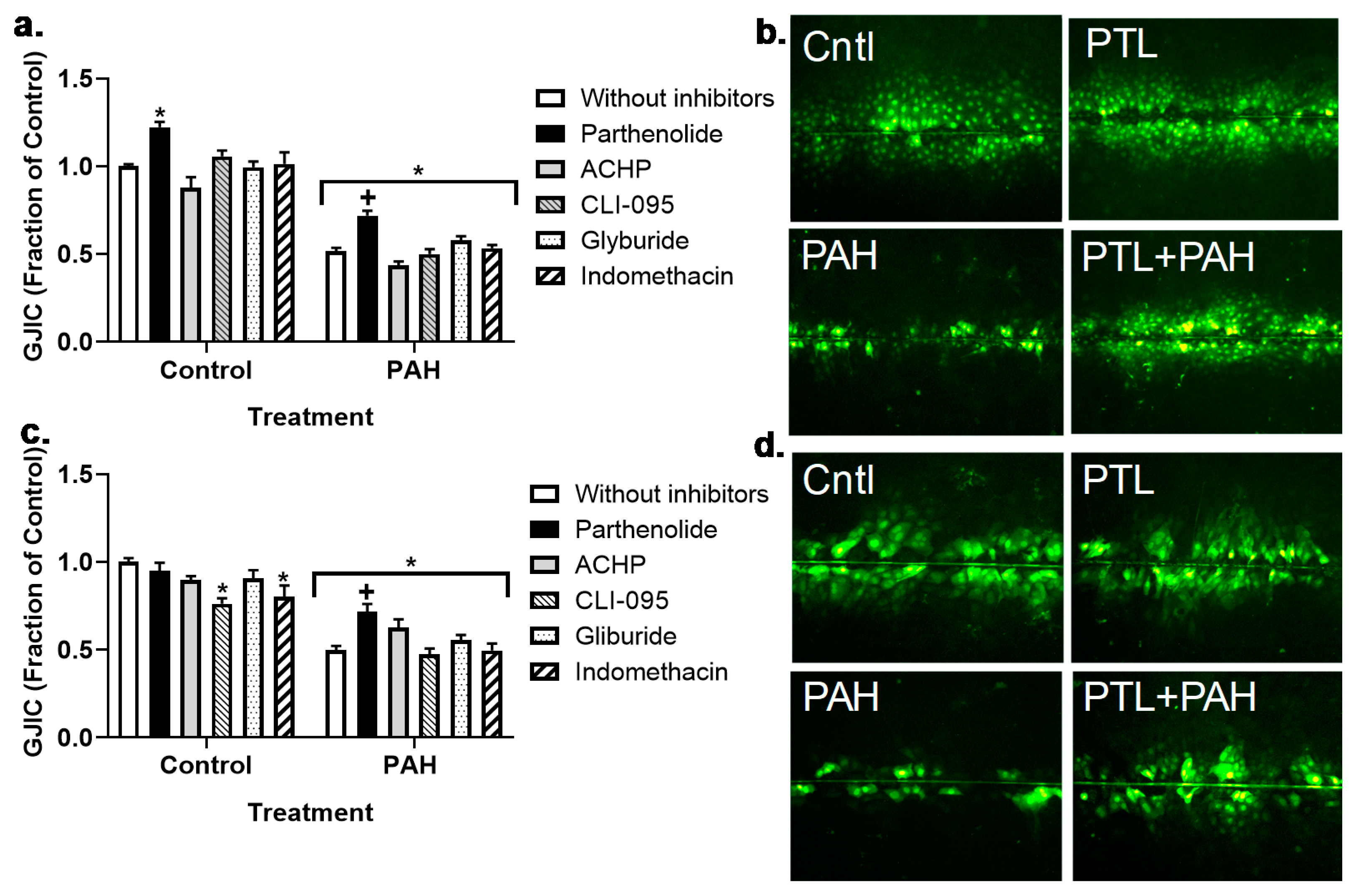
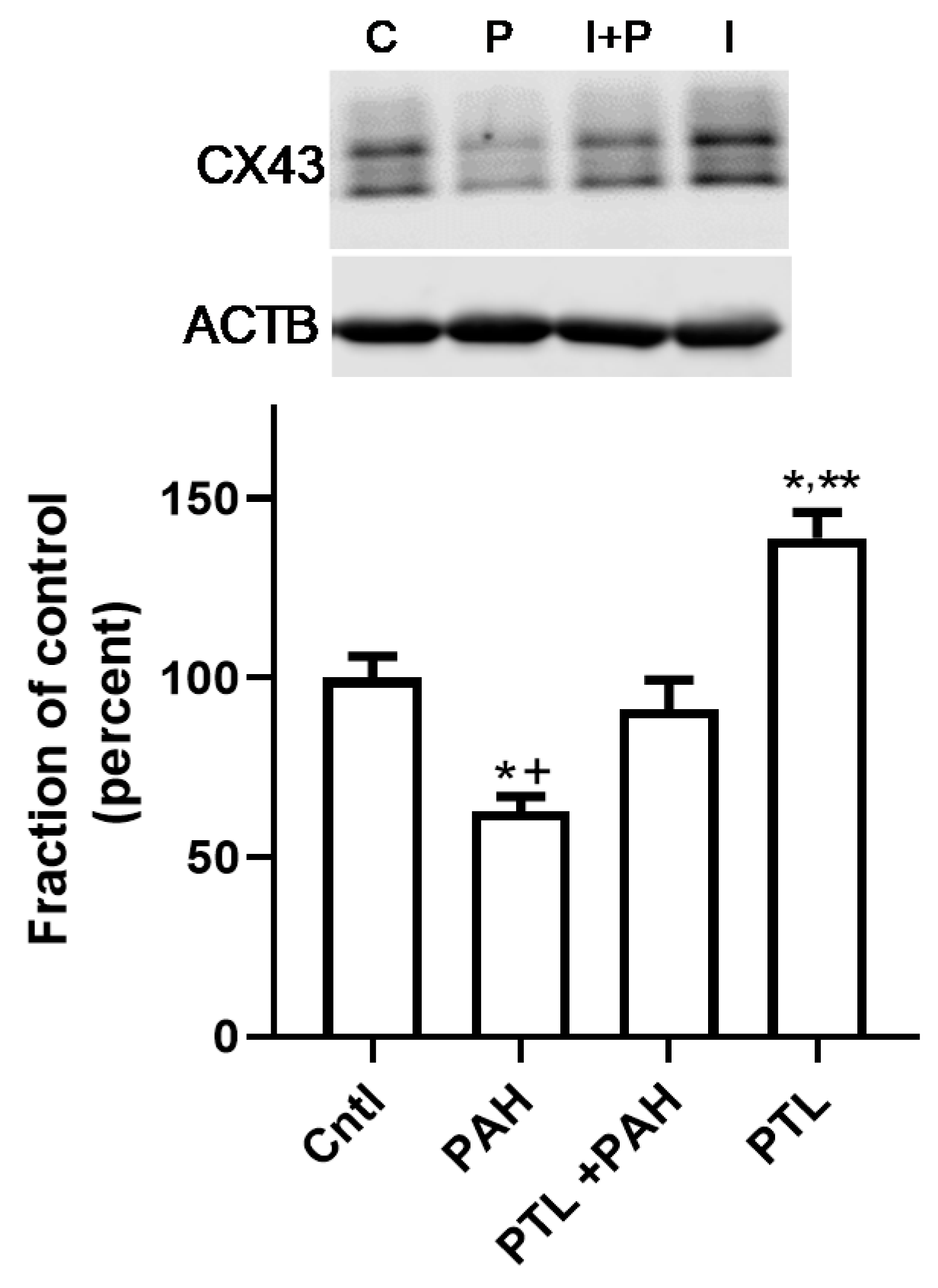
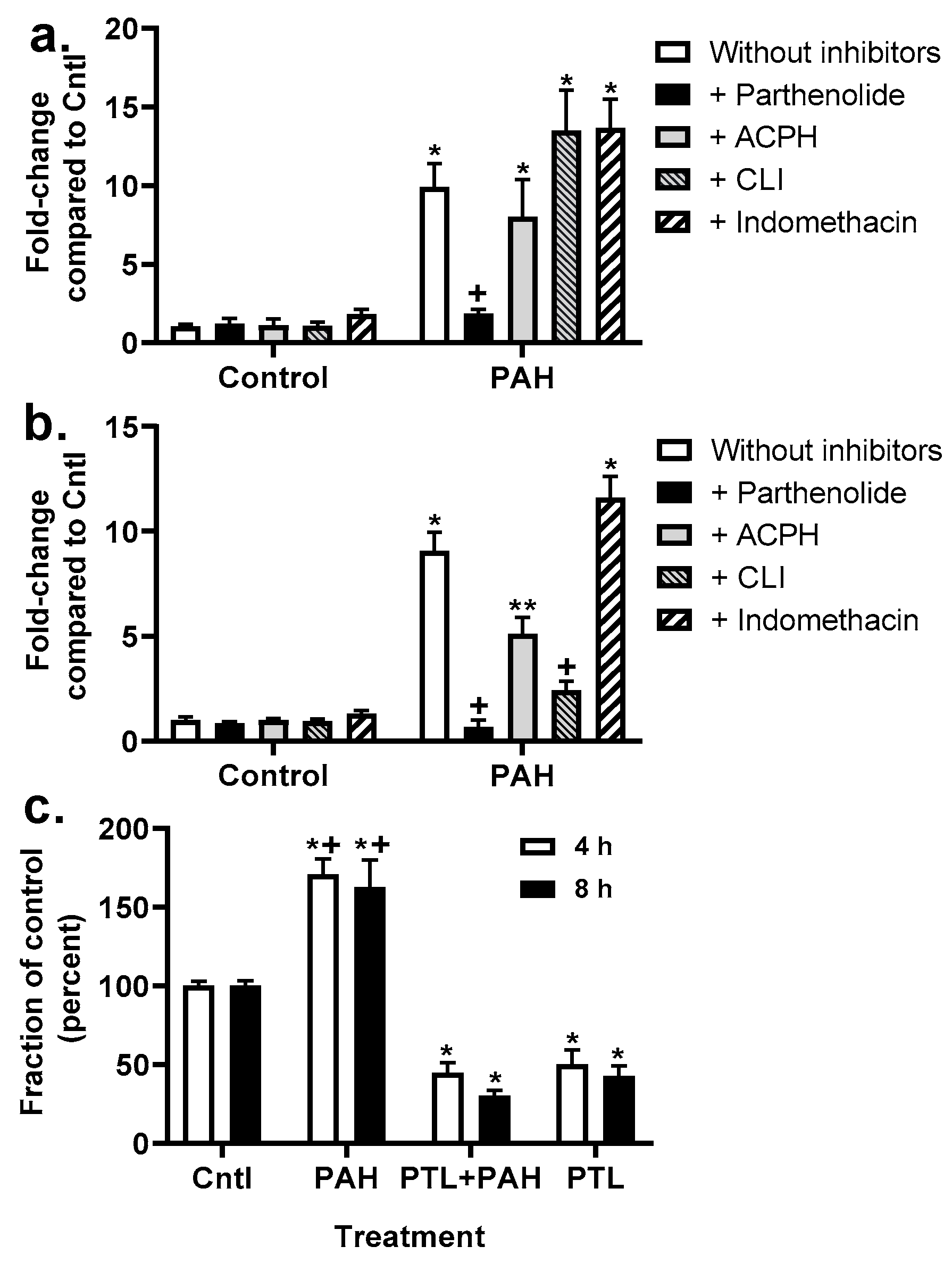
2.3. PAH-Induced Inhibition of Gap Junction Activity Is Prevented in Epithelial Cells in Response to a Pan-Inflammation Inhibitor
2.4. Chemokine Upregulation in Response to LMW PAHs: Inhibition with Anti-Inflammatory Compounds
2.5. Production of Cytokines and Chemokines in MHS Cells in Response to LMW PAHs: Effects of Anti-Inflammatory Compounds
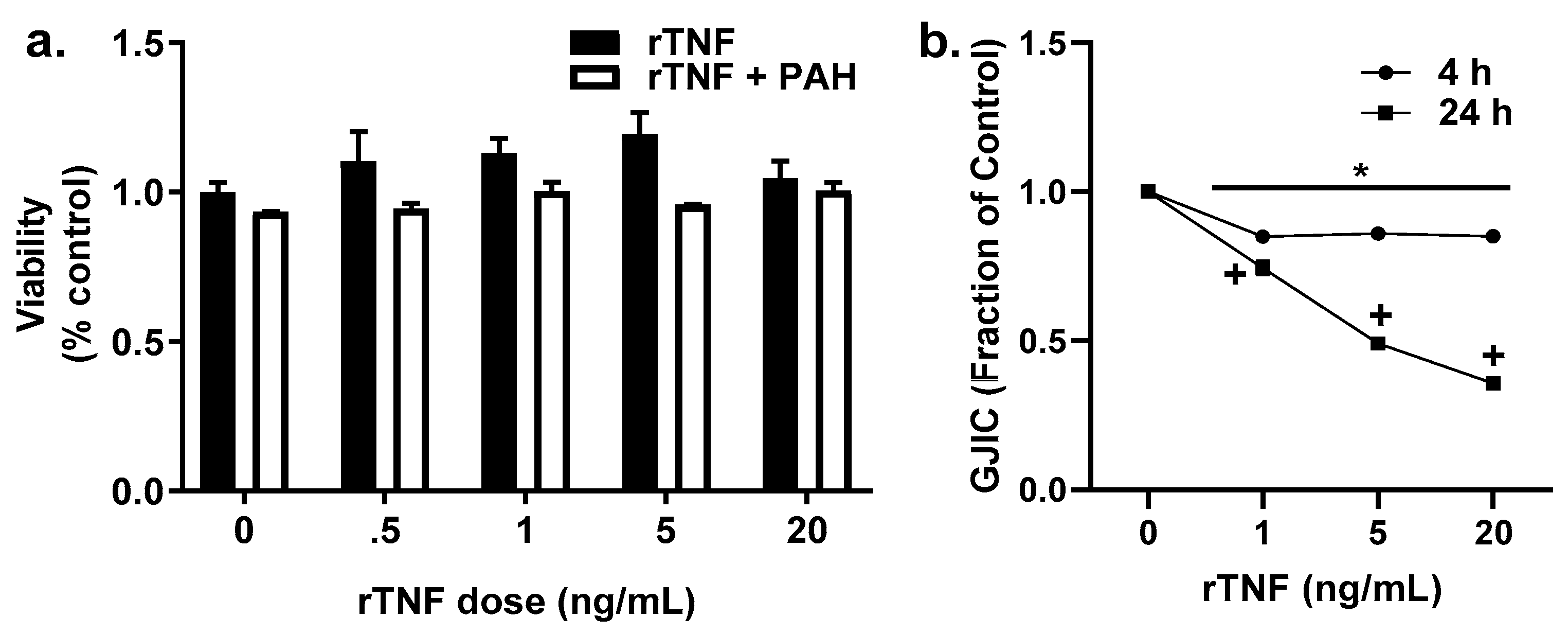
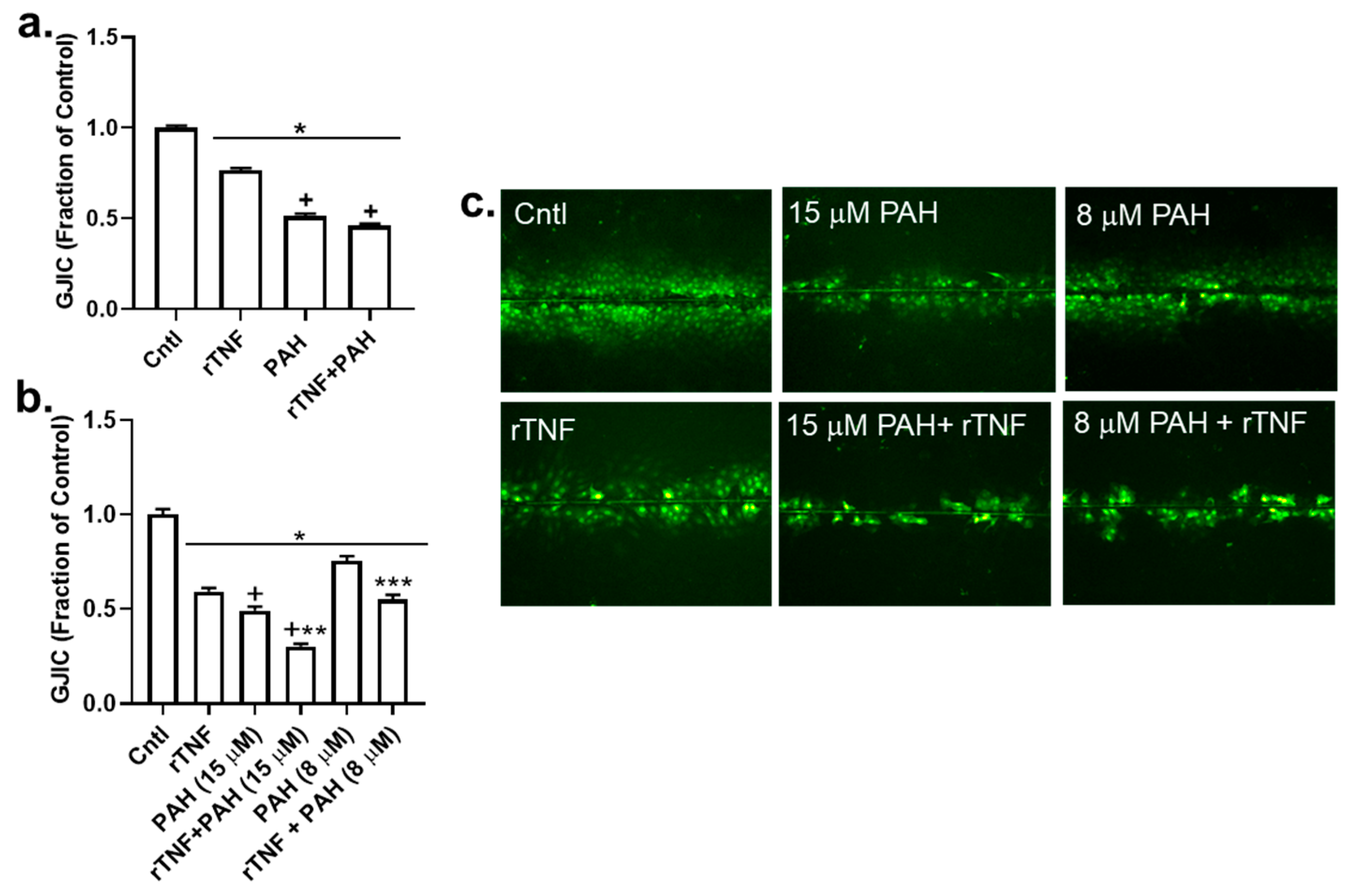
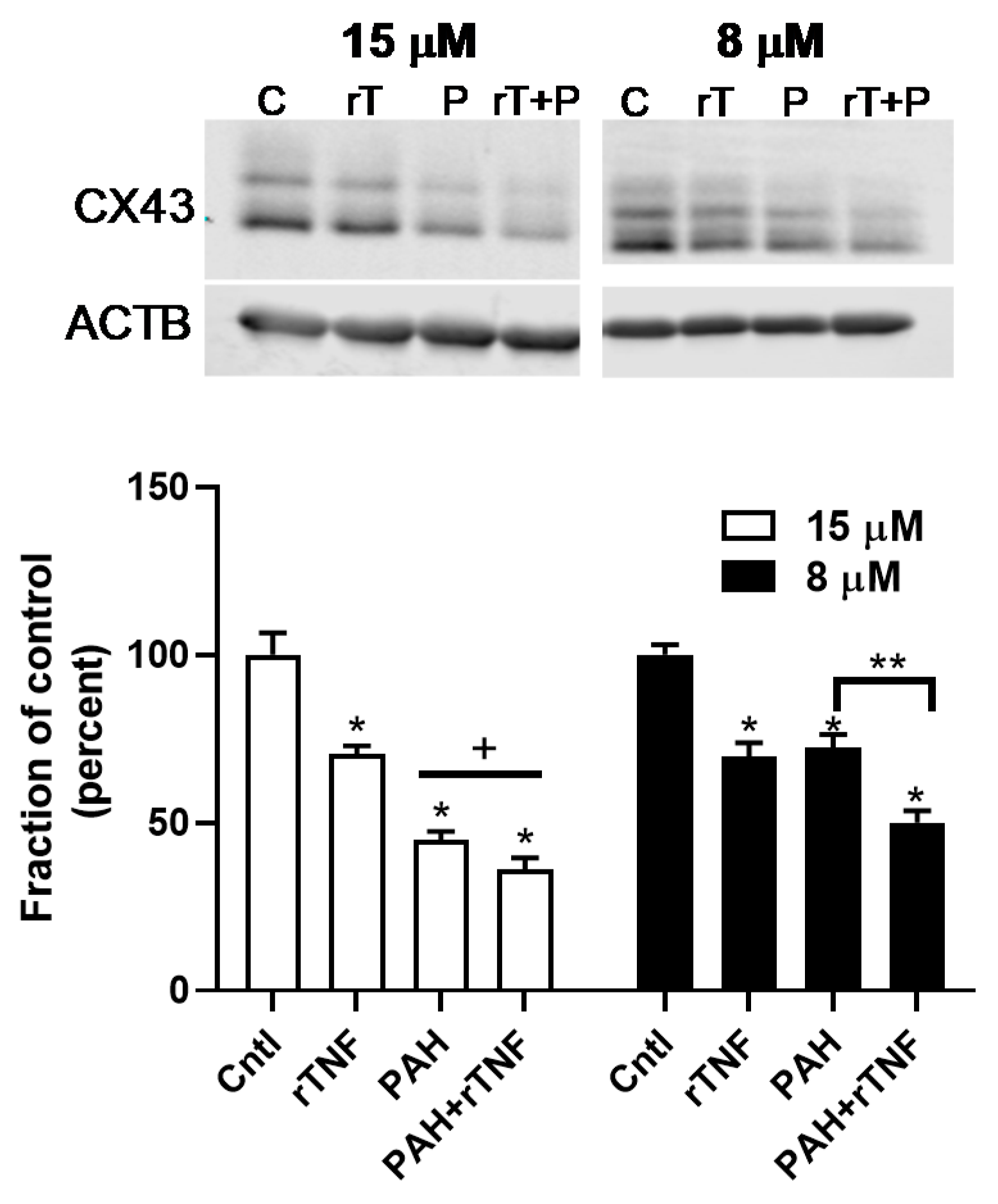
2.6. Recombinant TNF (rTNF) Elicits GJIC Inhibition Alone and in Response to Combinations of rTNF and PAHs
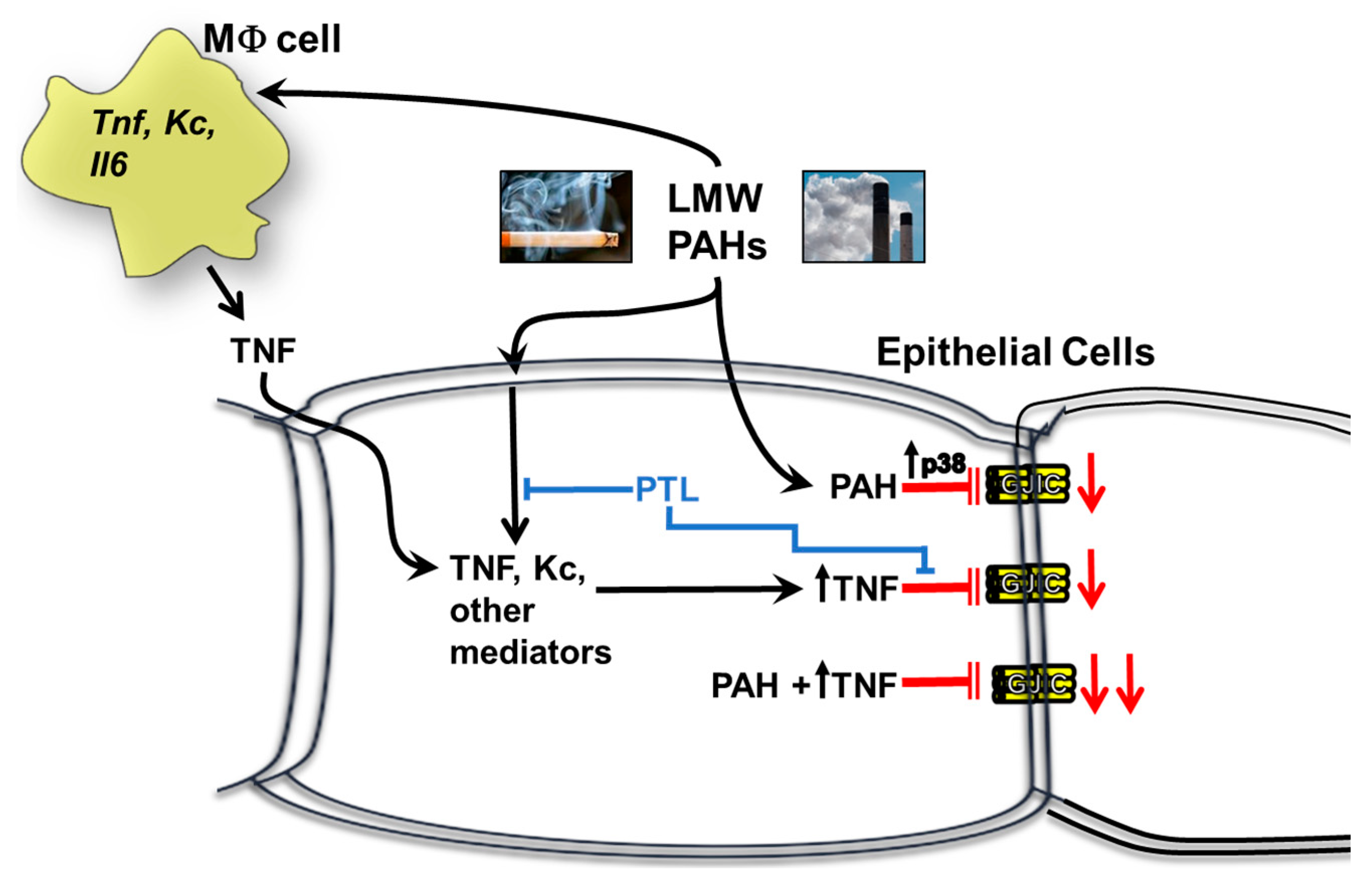
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Line Maintenance and Experimental Design
4.3. Cytotoxicity
4.4. Scalpel Loaded/Dye-Transfer Assays to Measure Gap Junctional Intercellular Communication (GJIC)
4.5. KC Measurement via ELISA
4.6. CX43 Protein Expression Using Immunoblots
4.7. Quantitative Reverse Transcriptase PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COX2 | cyclooxygenase 2 |
| CX | connexin |
| GJIC | gap junctional intercellular communication |
| HMW | high molecular weight |
| KC | keratinocyte chemoattractant; CXCl1 |
| MCP1 | monocyte chemoattractant protein 1 |
| LMW | low molecular weight |
| PAH | polycyclic aromatic hydrocarbon |
| TNF | tumor necrosis factor alpha |
| rTNF | recombinant TNF |
References
- Lee, H.L.; Hsieh, D.P.; Li, L.A. Polycyclic aromatic hydrocarbons in cigarette sidestream smoke particulates from a Taiwanese brand and their carcinogenic relevance. Chemosphere 2010. [Google Scholar] [CrossRef] [PubMed]
- Moir, D.; Rickert, W.S.; Levasseur, G.; Larose, Y.; Maertens, R.; White, P.; Desjardins, S. A comparison of mainstream and sidestream marijuana and tobacco cigarette smoke produced under two machine smoking conditions. Chem. Res. Toxicol. 2008, 21, 494–502. [Google Scholar] [CrossRef] [PubMed]
- ATSDR. Toxicology Profile for Polyaromatic Hydrocarbons; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Schick, S.F.; Farraro, K.F.; Perrino, C.; Sleiman, M.; van de Vossenberg, G.; Trinh, M.P.; Hammond, S.K.; Jenkins, B.M.; Balmes, J. Thirdhand cigarette smoke in an experimental chamber: Evidence of surface deposition of nicotine, nitrosamines and polycyclic aromatic hydrocarbons and de novo formation of NNK. Tob. Control 2013. [Google Scholar] [CrossRef] [PubMed]
- IARC. Outdoor Air Pollution; IARC: Lyon, France, 2016. [Google Scholar]
- World Health Organization. Ambient Air Pollution: A Global Assessment of Exposure and Burden of Disease; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- IARC. Personal Habits and Indoor Combustions; IARC: Lyon, France, 2012. [Google Scholar]
- D’Amato, G.; Baena-Cagnani, C.E.; Cecchi, L.; Annesi-Maesano, I.; Nunes, C.; Ansotegui, I.; D’Amato, M.; Liccardi, G.; Sofia, M.; Canonica, W.G. Climate change, air pollution and extreme events leading to increasing prevalence of allergic respiratory diseases. Multidiscip. Respir. Med. 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- IARC. Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures; IARC: Lyon, France, 2010. [Google Scholar]
- U.S.E.P.A. Polycyclic aromatic hydrocarbons, 15 listings. Rep. Carcinog. 2002, 10, 201–204. [Google Scholar]
- Severson, R.F.; Snook, M.E.; Higman, H.C.; Chortyk, O.T.; Akin, F.J. Isolation, identification, and quantification of polynuclear aromatic hydrocarbons in tobacco smoke. In Carcinogenesis—A comprehensive Survey. Vol. 1. Polynuclear Aromatic Hydrocarbons: Chemistry, Metabolism, and Carcinogenesis; Freudenthal, R.I., Jones, P.W., Eds.; Raven Press: New York, NY, USA, 1976; pp. 253–270. [Google Scholar]
- Hong, W.J.; Jia, H.; Ma, W.L.; Sinha, R.K.; Moon, H.B.; Nakata, H.; Minh, N.H.; Chi, K.H.; Li, W.L.; Kannan, K.; et al. Distribution, Fate, Inhalation Exposure and Lung Cancer Risk of Atmospheric Polycyclic Aromatic Hydrocarbons in Some Asian Countries. Environ. Sci. Technol. 2016, 50, 7163–7174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, Y. Polycyclic aromatic hydrocarbons contamination in surface soil of China: A review. Sci. Total Environ. 2017, 605–606, 1011–1020. [Google Scholar] [CrossRef]
- Marczynski, B.; Pesch, B.; Wilhelm, M.; Rossbach, B.; Preuss, R.; Hahn, J.U.; Rabstein, S.; Raulf-Heimsoth, M.; Seidel, A.; Rihs, H.P.; et al. Occupational exposure to polycyclic aromatic hydrocarbons and DNA damage by industry: A nationwide study in Germany. Arch. Toxicol. 2009, 83, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Kappler, M.; Straif, K.; Marczynski, B.; Preuss, R.; Rossbach, B.; Rihs, H.P.; Weiss, T.; Rabstein, S.; Pierl, C.; et al. Dose-response modeling of occupational exposure to polycyclic aromatic hydrocarbons with biomarkers of exposure and effect. Cancer Epidemiol. Biomark. Prev 2007, 16, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Talaska, G.; Thoroman, J.; Schuman, B.; Kafferlein, H.U. Biomarkers of polycyclic aromatic hydrocarbon exposure in European coke oven workers. Toxicol. Lett. 2014, 231, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Serdar, B.; Brindley, S.; Dooley, G.; Volckens, J.; Juarez-Colunga, E.; Gan, R. Short-term markers of DNA damage among roofers who work with hot asphalt. Environ. Health 2016, 15, 99. [Google Scholar] [CrossRef]
- Allan, S.E.; Smith, B.W.; Anderson, K.A. Impact of the deepwater horizon oil spill on bioavailable polycyclic aromatic hydrocarbons in Gulf of Mexico coastal waters. Environ. Sci. Technol. 2012, 46, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Bojes, H.K.; Pope, P.G. Characterization of EPA’s 16 priority pollutant polycyclic aromatic hydrocarbons (PAHs) in tank bottom solids and associated contaminated soils at oil exploration and production sites in Texas. Regul. Toxicol. Pharm. 2007, 47, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Fustinoni, S.; Campo, L.; Cirla, P.E.; Martinotti, I.; Buratti, M.; Longhi, O.; Foa, V.; Bertazzi, P. Dermal exposure to polycyclic aromatic hydrocarbons in asphalt workers. Occup. Environ. Med. 2010, 67, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J.; Banerjee, A.; Meena, R.K.; Kumari, K.M.; Lakhani, A. Characterization of polycyclic aromatic hydrocarbons in emissions of different mosquito coils. Bull. Environ. Contam. Toxicol. 2014, 92, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Lung, S.C.; Hu, S.C. Generation rates and emission factors of particulate matter and particle-bound polycyclic aromatic hydrocarbons of incense sticks. Chemosphere 2003, 50, 673–679. [Google Scholar] [CrossRef]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Szewczyńska, M.; Pośniak, M.; Dobrzyńska, E. Study on the individual PAHs content in ultrafine particles from solid fractions of diesel and biodiesel exhaust fumes. J. Chem. 2012, 2013, 1–10. [Google Scholar] [CrossRef]
- Bauer, A.K.; Malkinson, A.M.; Kleeberger, S.R. Susceptibility to neoplastic and non-neoplastic pulmonary diseases in mice: Genetic similarities. Am. J. Physiol. 2004, 287, L685–L703. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M.; Xu, Y. Epigenetic mechanisms of chemical carcinogenesis. Hum. Exp. Toxicol. 2000, 19, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Malkinson, A.M. Molecular comparison of human and mouse pulmonary adenocarcinomas. Exp. Lung Res. 1998, 24, 541–555. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Nahta, R.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Andrade-Vieira, R.; Bay, S.N.; Brown, D.G.; Calaf, G.M.; Castellino, R.C.; Cohen-Solal, K.A.; et al. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015, 36 (Suppl. 1), S2–S18. [Google Scholar] [CrossRef] [Green Version]
- Trosko, J.E. Commentary: Is the concept of “tumor promotion” a useful paradigm? Mol. Carcinog. 2001, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Chang, C.C. Oncogene and chemical inhibition of gap-junctional intercellular communication: Implications for teratogenesis and carcinogenesis. Prog. Clin. Biol. Res. 1986, 209B, 21–31. [Google Scholar] [PubMed]
- Trosko, J.E.; Chang, C.C.; Madhukar, B.V.; Dupont, E. Oncogenes, tumor suppressor genes and intercellular communication in the ‘Oncogeny as partially blocked ontogeny’ hypothesis. In New Frontiers in Cancer Causation; Iversen, O.H., Ed.; Taylor and Francis Publishers: Washington, DC, USA, 1993; pp. 181–197. [Google Scholar]
- Spath, C.; Schlegel, F.; Leontyev, S.; Mohr, F.W.; Dhein, S. Inverse Relationship between Tumor Proliferation Markers and Connexin Expression in a Malignant Cardiac Tumor Originating from Mesenchymal Stem Cell Engineered Tissue in a Rat in vivo Model. Front. Pharm. 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; Mackowiak, I.I.; Mori, C.M.; da Silva, T.C.; Oloris, S.C.; Garate, A.P.; Massironi, S.M.; Yamasaki, H.; et al. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 2004, 25, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.K.; Velmurugan, K.; Plottner, S.; Siegrist, K.J.; Romo, D.; Welge, P.; Bruning, T.; Xiong, K.N.; Kafferlein, H.U. Environmentally prevalent polycyclic aromatic hydrocarbons can elicit co-carcinogenic properties in an in vitro murine lung epithelial cell model. Arch. Toxicol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Osgood, R.S.; Upham, B.L.; Hill, T.; Helms, K.L.; Velmurugan, K.; Babica, P.; Bauer, A.K. Polycyclic aromatic hydrocarbon-induced signaling events relevant to inflammation and tumorigenesis in lung cells are dependent on molecular structure. PLoS ONE 2013, 8, e65150. [Google Scholar] [CrossRef] [PubMed]
- Osgood, R.S.; Upham, B.L.; Bushel, P.R.; Velmurugan, K.; Xiong, K.N.; Bauer, A.K. Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol. Sci. 2017, 157, 156–171. [Google Scholar] [CrossRef] [Green Version]
- Trovato-Salinaro, A.; Trovato-Salinaro, E.; Failla, M.; Mastruzzo, C.; Tomaselli, V.; Gili, E.; Crimi, N.; Condorelli, D.F.; Vancheri, C. Altered intercellular communication in lung fibroblast cultures from patients with idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 122. [Google Scholar] [CrossRef]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; da Silva, T.C.; Fukumasu, H.; Mori, C.M.; Yamasaki, H.; Dagli, M.L. Altered expression of connexins in urethane-induced mouse lung adenomas. Life Sci. 2006, 79, 2202–2208. [Google Scholar] [CrossRef]
- Siegrist, K.J.; Romo, D.; Upham, B.L.; Armstrong, M.; Quinn, K.; Vanderlinden, L.; Osgood, R.S.; Velmurugan, K.; Elie, M.; Manke, J.; et al. Early Mechanistic Events Induced by Low Molecular Weight Polycyclic Aromatic Hydrocarbons in Mouse Lung Epithelial Cells: A Role for Eicosanoid Signaling. Toxicol. Sci. 2019. [Google Scholar] [CrossRef]
- Guan, X.; Hardenbrook, J.; Fernstrom, M.J.; Chaudhuri, R.; Malkinson, A.M.; Ruch, R.J. Down-regulation by butylated hydroxytoluene of the number and function of gap junctions in epithelial cell lines derived from mouse lung and rat liver. Carcinogenesis 1995, 16, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N.; Koval, M. Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication. Antioxid. Redox Signal. 2009, 11, 355–367. [Google Scholar] [CrossRef]
- Chanson, M.; Berclaz, P.Y.; Scerri, I.; Dudez, T.; Wernke-Dollries, K.; Pizurki, L.; Pavirani, A.; Fiedler, M.A.; Suter, S. Regulation of gap junctional communication by a pro-inflammatory cytokine in cystic fibrosis transmembrane conductance regulator-expressing but not cystic fibrosis airway cells. Am. J. Pathol. 2001, 158, 1775–1784. [Google Scholar] [CrossRef]
- Vikis, H.G.; Gelman, A.E.; Franklin, A.; Stein, L.; Rymaszewski, A.; Zhu, J.; Liu, P.; Tichelaar, J.W.; Krupnick, A.S.; You, M. Neutrophils are required for 3-methylcholanthrene-initiated, butylated hydroxytoluene-promoted lung carcinogenesis. Mol. Carcinog. 2012, 51, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Cumpian, A.M.; Caetano, M.S.; Ochoa, C.E.; De la Garza, M.M.; Lapid, D.J.; Mirabolfathinejad, S.G.; Dickey, B.F.; Zhou, Q.; Moghaddam, S.J. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol. Cancer 2013, 12, 154. [Google Scholar] [CrossRef]
- Gong, L.; da Silva Caetano, M.; Cumpian, A.M.; Daliri, S.; Garza Flores, A.; Chang, S.H.; Ochoa, C.E.; Evans, C.M.; Yu, Z.; Moghaddam, S.J. Tumor necrosis factor links chronic obstructive pulmonary disease and K-ras mutant lung cancer through induction of an immunosuppressive pro-tumor microenvironment. Oncoimmunology 2016, 5, e1229724. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.M.; Xiong, K.N.; Velmurugan, K.; Xiong, J.; Osgood, R.S.; Bauer, A.K. Differential innate immune cell signatures and effects regulated by toll-like receptor 4 during murine lung tumor promotion. Exp. Lung Res. 2016, 42, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.M.; Dwyer-Nield, L.D.; Malkinson, A.M. Stimulation of neoplastic mouse lung cell proliferation by alveolar macrophage-derived, insulin-like growth factor-1 can be blocked by inhibiting MEK and PI3K activation. Mol. Cancer 2011, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Upham, B.L.; Blaha, L.; Babica, P.; Park, J.S.; Sovadinova, I.; Pudrith, C.; Rummel, A.M.; Weis, L.M.; Sai, K.; Tithof, P.K.; et al. Tumor promoting properties of a cigarette smoke prevalent polycyclic aromatic hydrocarbon as indicated by the inhibition of gap junctional intercellular communication via phosphatidylcholine-specific phospholipase C. Cancer Sci. 2008, 99, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Cho, H.Y.; Morgan, D.L.; Bauer, A.K.; Kleeberger, S.R. Signal transduction pathways of tumor necrosis factor--mediated lung injury induced by ozone in mice. Am. J. Respir. Crit. Care Med. 2007, 175, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Kabatkova, M.; Svobodova, J.; Pencikova, K.; Mohatad, D.S.; Smerdova, L.; Kozubik, A.; Machala, M.; Vondracek, J. Interactive effects of inflammatory cytokine and abundant low-molecular-weight PAHs on inhibition of gap junctional intercellular communication, disruption of cell proliferation control, and the AhR-dependent transcription. Toxicol. Lett. 2015, 232, 113–121. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Eastman, J.; Berger, M.; Bonfield, T.L. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflamm. (Lond.) 2011, 8, 26. [Google Scholar] [CrossRef]
- Ii, M.; Matsunaga, N.; Hazeki, K.; Nakamura, K.; Takashima, K.; Seya, T.; Hazeki, O.; Kitazaki, T.; Iizawa, Y. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharm. 2006, 69, 1288–1295. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Bernert, H.; Sekikawa, K.; Radcliffe, R.A.; Iraqi, F.; You, M.; Malkinson, A.M. Tnfa and Il-10 deficiencies have contrasting effects on lung tumor susceptibility: Gender-dependent modulation of IL-10 haploinsufficiency. Mol. Carcinog. 2003, 38, 117–123. [Google Scholar] [CrossRef]
- Smerdova, L.; Smerdova, J.; Kabatkova, M.; Kohoutek, J.; Blazek, D.; Machala, M.; Vondracek, J. Upregulation of CYP1B1 expression by inflammatory cytokines is mediated by the p38 MAP kinase signal transduction pathway. Carcinogenesis 2014, 35, 2534–2543. [Google Scholar] [CrossRef] [Green Version]
- Umannova, L.; Machala, M.; Topinka, J.; Schmuczerova, J.; Krcmar, P.; Neca, J.; Sujanova, K.; Kozubik, A.; Vondracek, J. Benzo[a]pyrene and tumor necrosis factor-alpha coordinately increase genotoxic damage and the production of proinflammatory mediators in alveolar epithelial type II cells. Toxicol. Lett. 2011, 206, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Weber, W.J.; Rummel, A.M.; Trosko, J.E.; Upham, B.L. Epigenetic Toxicity of a Mixture of Polycyclic Aromatic Hydrocarbonson Gap Junctional Intercellular Communication Before and After Biodegradation. Environ. Sci. Technol. 1999, 33, 1044–1050. [Google Scholar] [CrossRef]
- Tai, M.H.; Upham, B.L.; Olson, L.K.; Tsao, M.S.; Reed, D.N., Jr.; Trosko, J.E. Cigarette smoke components inhibited intercellular communication and differentiation in human pancreatic ductal epithelial cells. Int. J. Cancer 2007, 120, 1855–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koffler, L.; Roshong, S.; Kyu Park, I.; Cesen-Cummings, K.; Thompson, D.C.; Dwyer-Nield, L.D.; Rice, P.; Mamay, C.; Malkinson, A.M.; Ruch, R.J. Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1)after forced connexin expression in lung and liver carcinoma cells. J. Cell. Biochem. 2000, 79, 347–354. [Google Scholar] [CrossRef]
- Chatterjee, S.; Baeter, S.; Bhattacharya, J. Endothelial and epithelial signaling in the lung. Am. J. Physiol. 2007, 293, L517–L519. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, K.; Ichimura, H.; Monma, E.; Lindert, J.; Quadri, S.; Issekutz, A.; Bhattacharya, J. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J. Clin. Investig. 2006, 116, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, V.; Dhar, D.K.; Warner, A.E.; Vivien Li, W.Y.; Amiri, A.F.; Bright, B.; Mookerjee, R.P.; Davies, N.A.; Becker, D.L.; Jalan, R. Importance of Connexin-43 based gap junction in cirrhosis and acute-on-chronic liver failure. J. Hepatol. 2013, 58, 1194–1200. [Google Scholar] [CrossRef]
- Dwyer-Nield, L.D.; Srebernak, M.C.; Barrett, B.S.; Ahn, J.; Cosper, P.; Meyer, A.M.; Kisley, L.R.; Bauer, A.K.; Thompson, D.C.; Malkinson, A.M. Cytokines differentially regulate the synthesis of prostanoid and nitric oxide mediators in tumorigenic versus non-tumorigenic mouse lung epithelial cell lines. Carcinogenesis 2005, 26, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Jiang, Q.; Willette-Brown, J.; Xi, S.; Zhu, F.; Burkett, S.; Back, T.; Song, N.Y.; Datla, M.; Sun, Z.; et al. The pivotal role of IKKalpha in the development of spontaneous lung squamous cell carcinomas. Cancer Cell 2013, 23, 527–540. [Google Scholar] [CrossRef]
- Bauer, A.K.; Rondini, E.A.; Hummel, K.A.; Degraff, L.M.; Walker, C.; Jedlicka, A.E.; Kleeberger, S.R. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: A role for heat-shock protein 70. Environ. Health Perspect. 2011, 119, 1091–1097. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Qureshi, S.; Homer, R.; Medzhitov, R.; Noble, P.W.; Lee, P.J. Cutting edge: TLR4 deficiency confers susceptibility to lethal oxidant lung injury. J. Immunol. 2005, 175, 4834–4838. [Google Scholar] [CrossRef]
- Bauer, A.K.; Upham, B.L.; Rondini, E.A.; Tennis, M.A.; Velmuragan, K.; Wiese, D. Toll-like receptor expression in human non-small cell lung carcinoma: Potential prognostic indicators of disease. Oncotarget 2017, 8, 91860–91875. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.K.; Jeng, C.J.; Wang, H.S.; Wang, S.H.; Wu, J.C. Lipopolysaccharide induces degradation of connexin43 in rat astrocytes via the ubiquitin-proteasome proteolytic pathway. PLoS ONE 2013, 8, e79350. [Google Scholar] [CrossRef] [PubMed]
- Hill, T., 3rd; Osgood, R.S.; Velmurugan, K.; Alexander, C.M.; Upham, B.L.; Bauer, A.K. Bronchoalveolar Lavage Fluid Utilized Ex Vivo to Validate In Vivo Findings: Inhibition of Gap Junction Activity in Lung Tumor Promotion is Toll-Like Receptor 4-Dependent. J. Mol. Biomark. Diagn. 2013, 5. [Google Scholar] [CrossRef]
- Malkinson, A.M.; Dwyer-Nield, L.D.; Rice, P.L.; Dinsdale, D. Mouse lung epithelial cell lines—Tools for the study of differentiation and the neoplastic phenotype. Toxicology 1997, 123, 53–100. [Google Scholar] [CrossRef]
- Bentel, J.M.; Lykke, A.W.; Smith, G.J. Cloned murine non-malignant, spontaneously transformed and chemical tumour-derived cell lines related to the type 2 pneumocyte. Cell Biol. Int. Rep. 1989, 13, 729–738. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Song, H.; Huang, C.; Yao, E.; Gacayan, R.; Xu, S.M.; Chuang, P.T. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE 2012, 7, e53817. [Google Scholar] [CrossRef]
- Mbawuike, I.N.; Herscowitz, H.B. MH-S, a murine alveolar macrophage cell line: Morphological, cytochemical, and functional characteristics. J. Leukoc. Biol. 1989, 46, 119–127. [Google Scholar] [CrossRef]
- Upham, B.L. Role of integrative signaling through gap junctions in toxicology. Curr. Protoc. Toxicol. 2011. [Google Scholar] [CrossRef]
- Bauer, A.K.; Velmurugan, K.; Xiong, K.N.; Alexander, C.M.; Xiong, J.; Brooks, R. Epiregulin is required for lung tumor promotion in a murine two-stage carcinogenesis model. Mol. Carcinog. 2017, 56, 94–105. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAH Dose * (μM) | Cox2 | Tnf | Kc | Mcp1 | Il6 | Il1β | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ave | SEM | Ave | SEM | Ave | SEM | Ave | SEM | Ave | SEM | Ave | SEM | |
| 0 | 1.02 | 0.13 | 1.01 | 0.03 | 1.01 | 0.11 | 1.00 | 0.06 | 1.04 | 0.19 | 1.00 | 0.07 |
| 5 | 1.70 | 0.15 | 1.51 | 0.11 | 1.37 | 0.35 | 1.22 | 0.07 | 1.33 | 0.38 | 2.01 | 0.35 |
| 10 | 1.02 | 0.09 | 1.41 | 0.19 | 1.28 | 0.20 | 1.55 | 0.42 | 1.72 | 0.35 | 2.79 | 0.30 ** |
| 15 | 2.30 | 0.44 ** | 2.46 | 0.30 ** | 1.81 | 0.32 | 1.97 | 0.16 | 1.73 | 0.12 | 5.42 | 0.16 ** |
| 20 | 2.18 | 0.11 ** | 2.78 | 0.25 ** | 3.09 | 1.13 ** | 2.02 | 0.31 | 3.20 | 0.67 ** | 6.26 | 0.16 ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romo, D.; Velmurugan, K.; Upham, B.L.; Dwyer-Nield, L.D.; Bauer, A.K. Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model. Cancers 2019, 11, 572. https://doi.org/10.3390/cancers11040572
Romo D, Velmurugan K, Upham BL, Dwyer-Nield LD, Bauer AK. Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model. Cancers. 2019; 11(4):572. https://doi.org/10.3390/cancers11040572
Chicago/Turabian StyleRomo, Deedee, Kalpana Velmurugan, Brad L. Upham, Lori D. Dwyer-Nield, and Alison K. Bauer. 2019. "Dysregulation of Gap Junction Function and Cytokine Production in Response to Non-Genotoxic Polycyclic Aromatic Hydrocarbons in an In Vitro Lung Cell Model" Cancers 11, no. 4: 572. https://doi.org/10.3390/cancers11040572