Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance


 ,
, 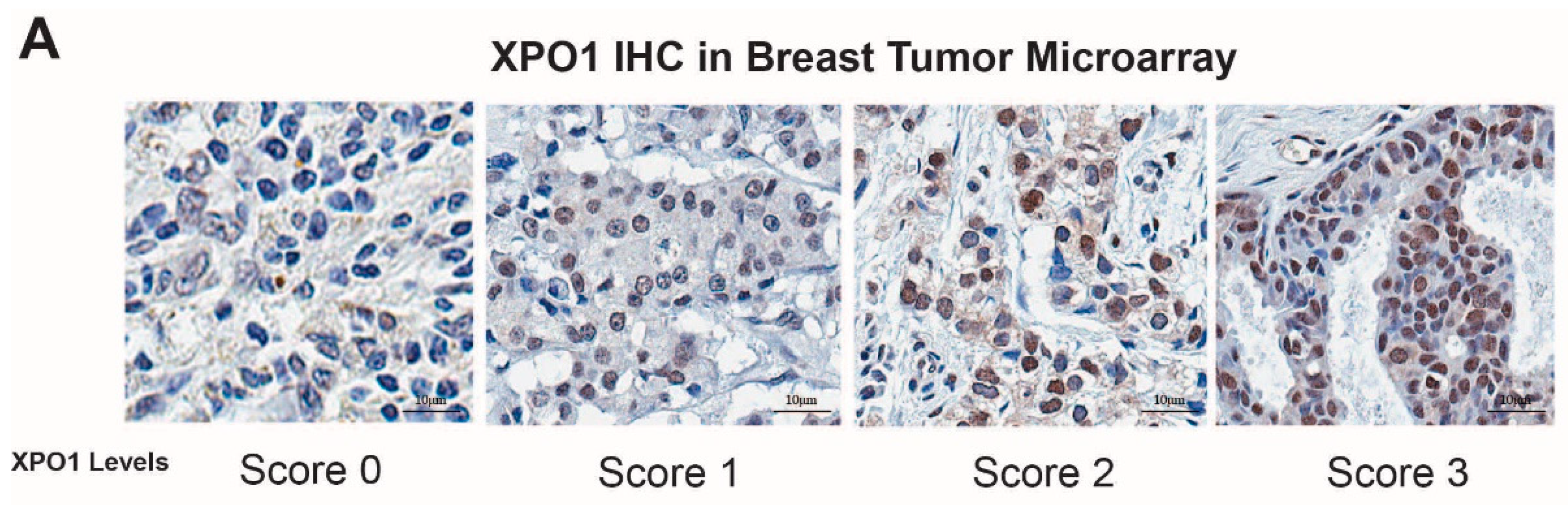{kind=link}
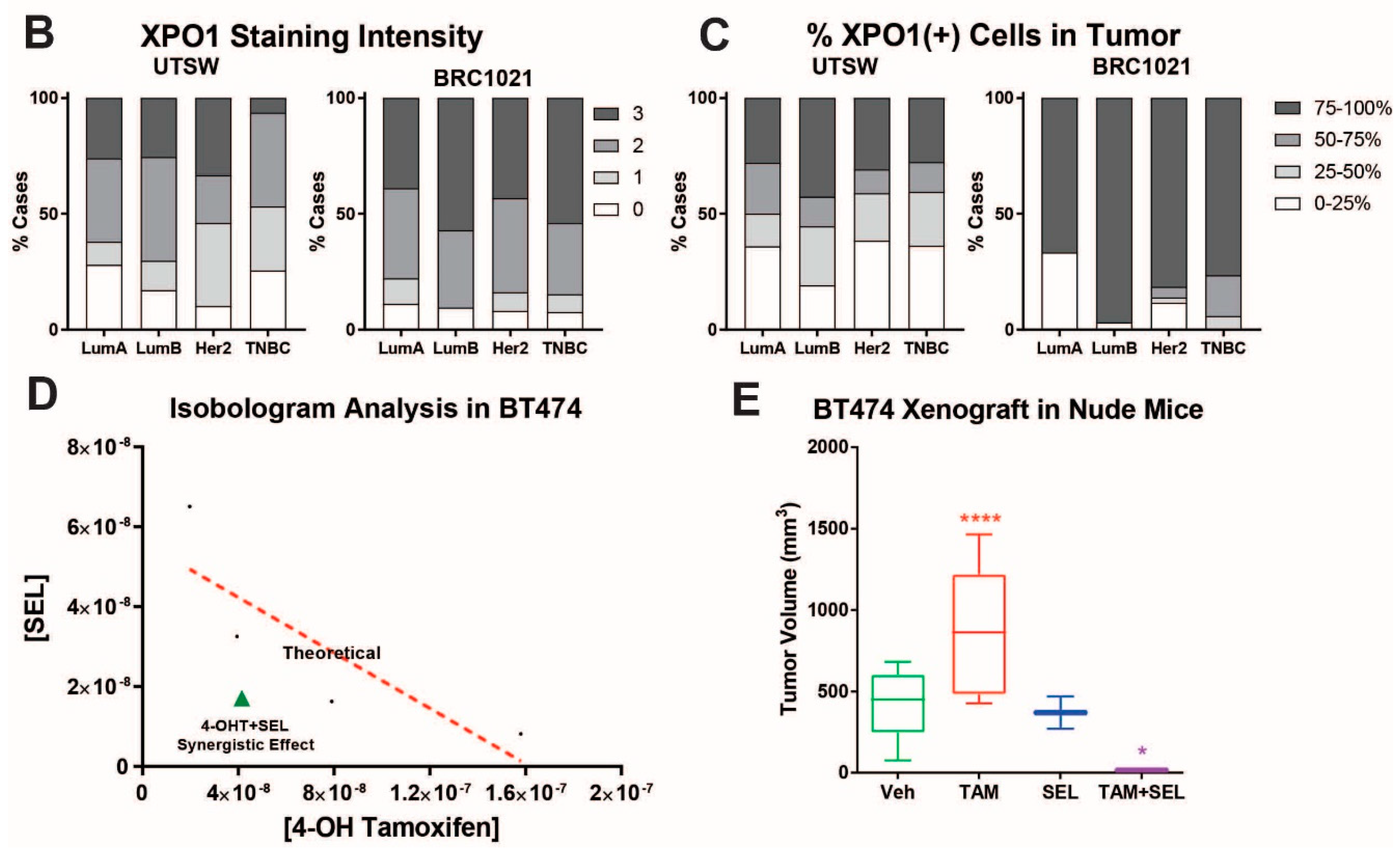{kind=link}
{kind=link}
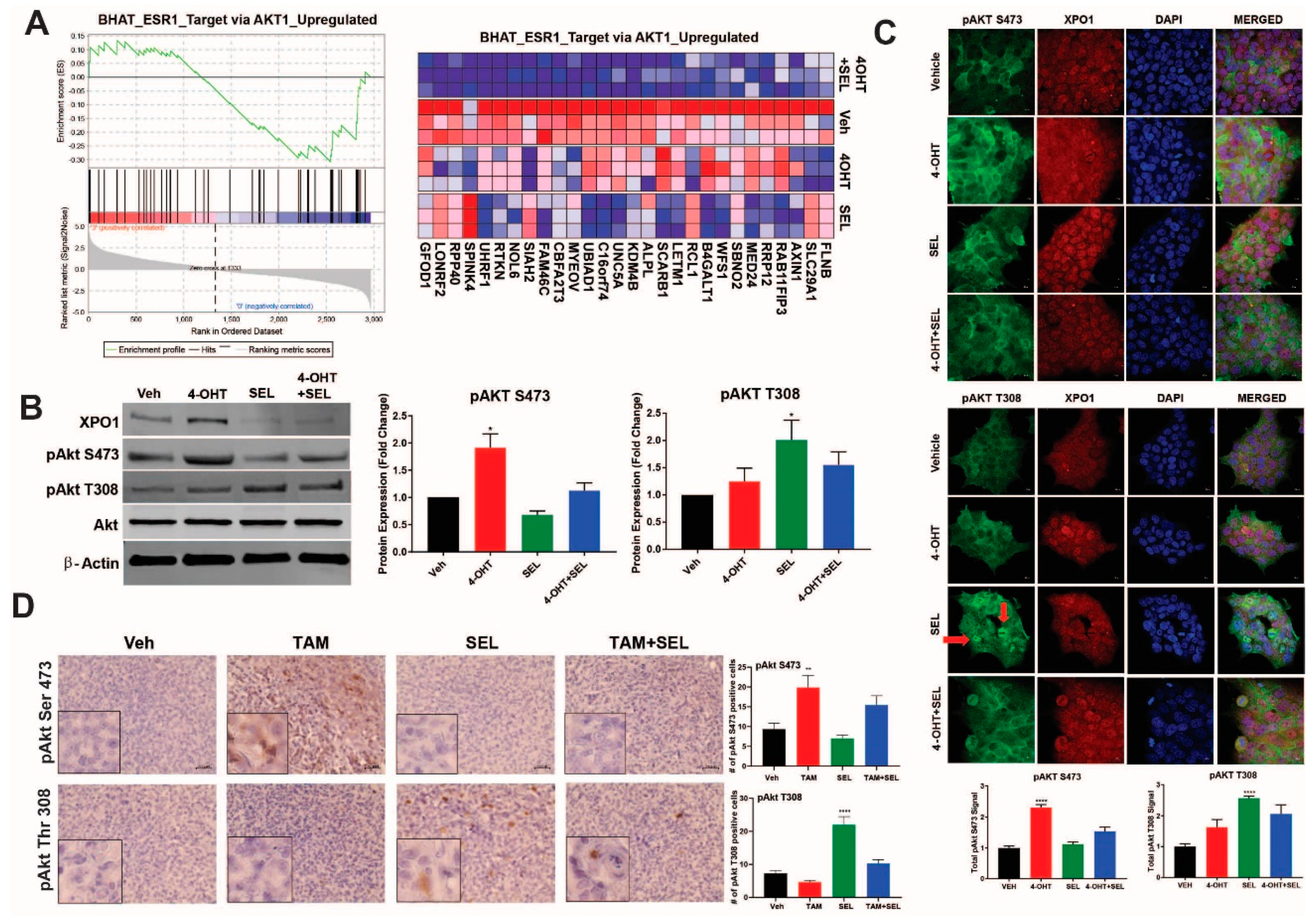{kind=link}
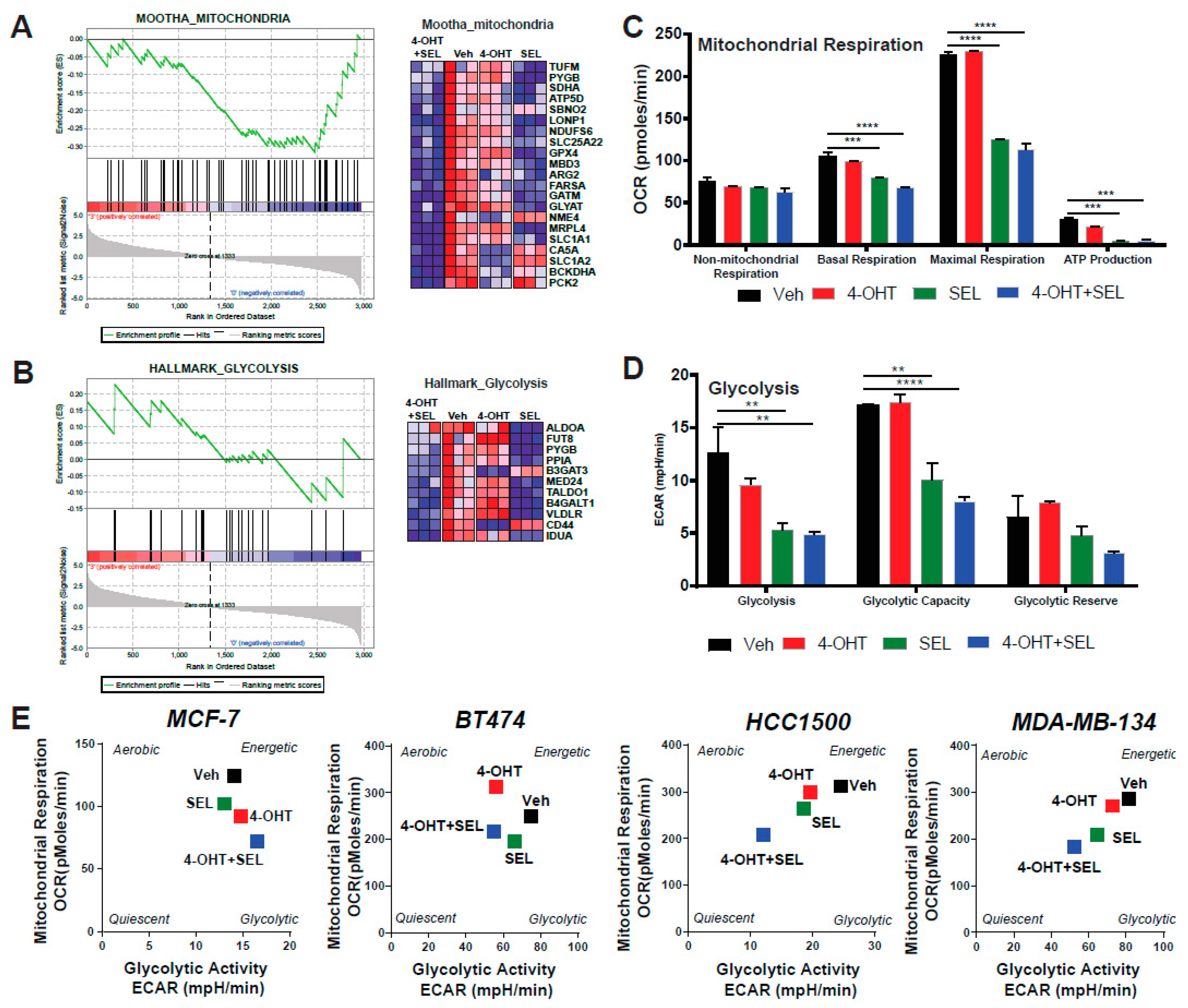{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Ligand Treatments
2.2. RNA Sequencing Analysis
2.3. In Vivo Xenograft Study, Immunohistochemistry Staining (IHC) and Data Analysis
2.4. Western Blot Analysis in Cell Lines
2.5. Seahorse Metabolic Profiling Assays
2.6. Caspase Colorimetric Protease Assay
2.7. Autophagy Assay
2.8. Cell Cycle Analysis
2.9. Statistical Analyses
2.10. Availability of Data and Materials
3. Results
3.1. 4-OHT+SEL Treatment Synergistically Sustains Tumor Regression in Endocrine Resistant Luminal B Subtype Breast Cancer Cells
3.2. 4-OHT+SEL Combination Treatment Causes Gene Expression Changes Distinct from 4-OHT and SEL Treatments Alone in TAM-Resistant Cell Lines
3.3. XPO1 Inhibition Modulates Differential Akt Phosphorylation in TAM-Resistant Cells and Tumor Xenografts
3.4. ERα-XPO1 Targeting Changes the Metabolic Phenotype of Breast Cancer Cells from An Energetic to A Quiescent Profile
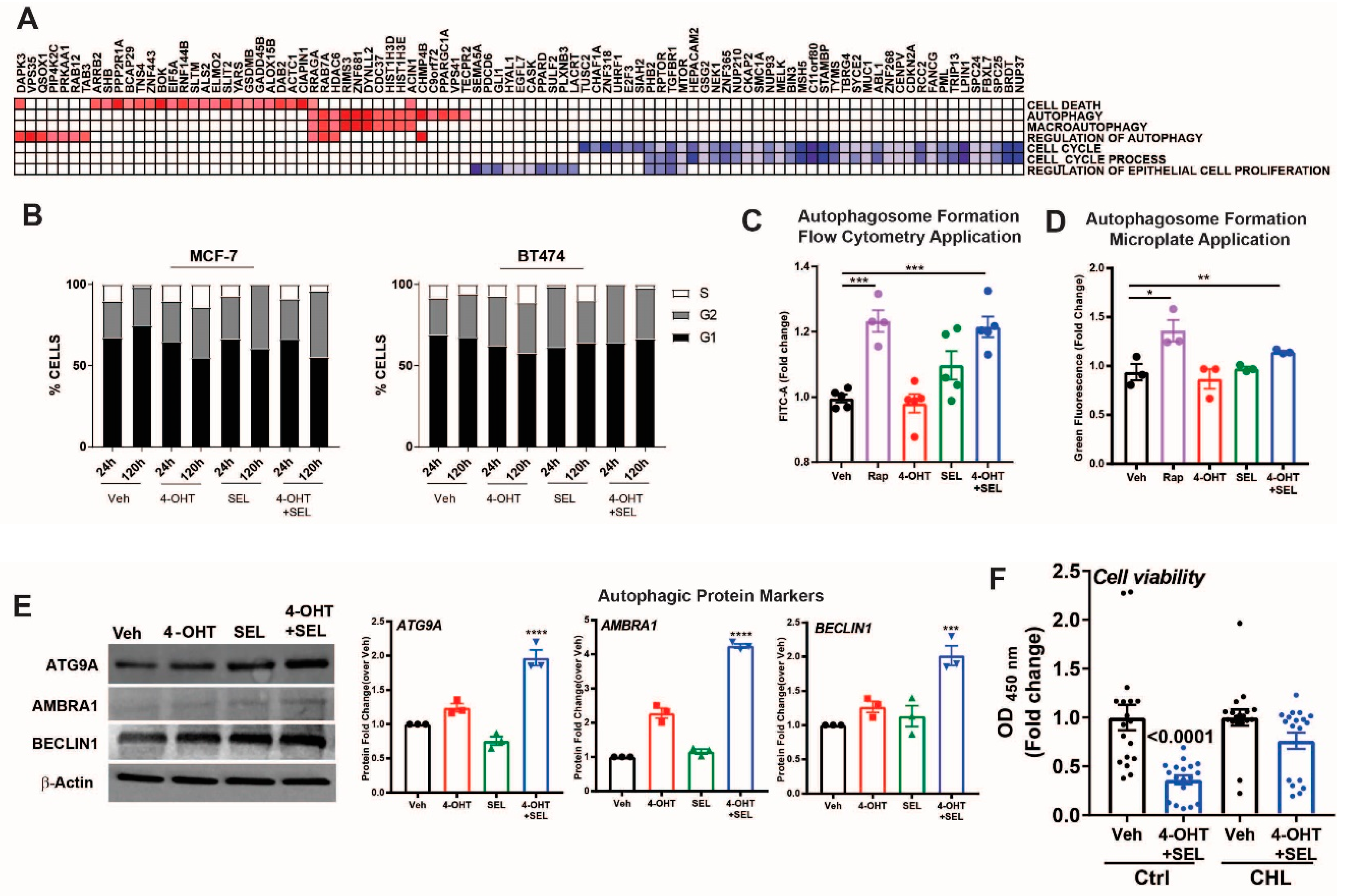
3.5. ERα-XPO1 Targeting Induces Autophagic Cell Death
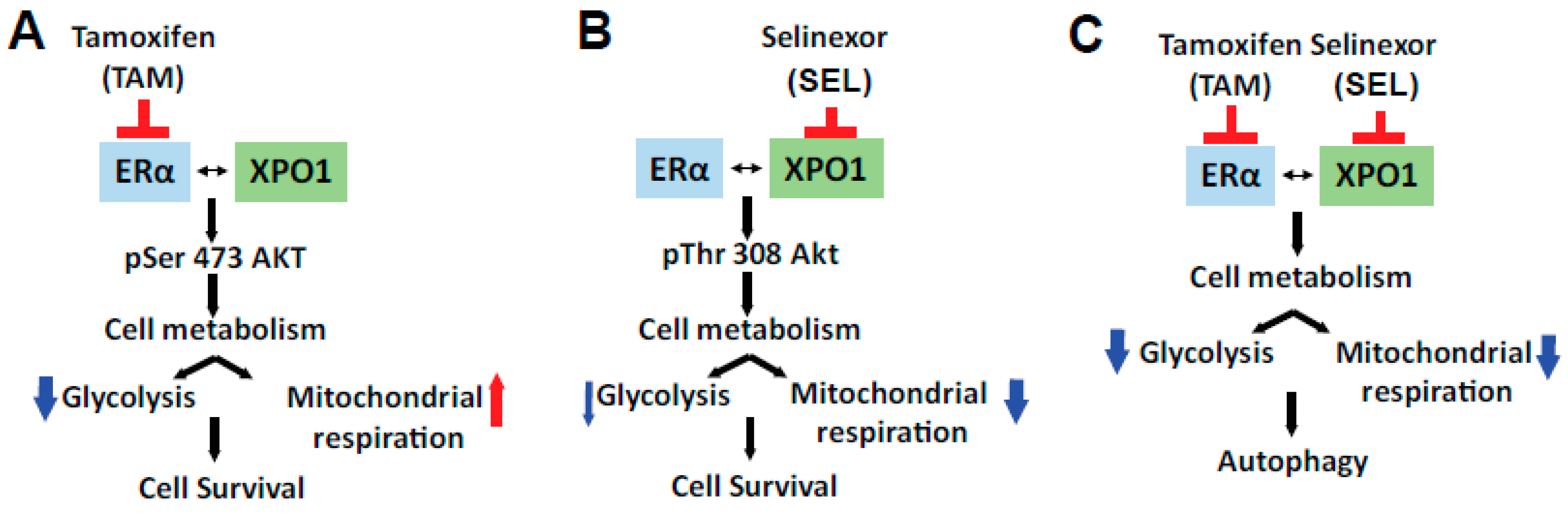
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Katzenellenbogen, B.S.; Frasor, J. Therapeutic targeting in the estrogen receptor hormonal pathway. Semin. Oncol. 2004, 31, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor–Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef]
- Yu, K.-D.; Wu, J.; Shen, Z.-Z.; Shao, Z.-M. Hazard of Breast Cancer-Specific Mortality among Women with Estrogen Receptor-Positive Breast Cancer after Five Years from Diagnosis: Implication for Extended Endocrine Therapy. J. Clin. Endocrinol. Metab. 2012, 97, E2201–E2209. [Google Scholar] [CrossRef] [PubMed]
- Jatoi, I.; Anderson, W.F.; Jeong, J.-H.; Redmond, C.K. Breast Cancer Adjuvant Therapy: Time to Consider Its Time-Dependent Effects. J. Clin. Oncol. 2011, 29, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Temin, S.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.E.; Giordano, S.H.; Hudis, C.A.; Rowden, D.; Solky, A.J.; et al. Adjuvant Endocrine Therapy for Women with Hormone Receptor–Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2014, 32, 2255–2269. [Google Scholar] [CrossRef]
- Colleoni, M.; Sun, Z.; Price, K.N.; Karlsson, P.; Forbes, J.F.; Thurlimann, B.; Gianni, L.; Castiglione, M.; Gelber, R.D.; Coates, A.S.; et al. Annual Hazard Rates of Recurrence for Breast Cancer During 24 Years of Follow-Up: Results from the International Breast Cancer Study Group Trials I to V. J. Clin. Oncol. 2016, 34, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Jatoi, I.; Chen, B.E.; Anderson, W.F.; Rosenberg, P.S. Breast cancer mortality trends in the United States according to estrogen receptor status and age at diagnosis. J. Clin. Oncol. 2007, 25, 1683–1690. [Google Scholar] [CrossRef]
- Kulkoyluoglu, E.; Madak-Erdogan, Z. Nuclear and extranuclear-initiated estrogen receptor signaling crosstalk and endocrine resistance in breast cancer. Steroids 2016, 114, 41–47. [Google Scholar] [CrossRef]
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62. [Google Scholar]
- Wrobel, K.; Zhao, Y.C.; Kulkoyluoglu, E.; Chen, K.L.; Hieronymi, K.; Holloway, J.; Li, S.; Ray, T.; Ray, P.S.; Landesman, Y.; et al. ERα-XPO1 Cross Talk Controls Tamoxifen Sensitivity in Tumors by Altering ERK5 Cellular Localization. Mol. Endocrinol. 2016, 30, 1029–1045. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.L.; Grant, S.; Shain, K.H.; Dalton, W.S.; Dai, Y.; Meads, M.; Baz, R.; Kauffman, M.; Shacham, S.; et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J. Hematol. Oncol. 2016, 9, 73. [Google Scholar] [CrossRef]
- Gravina, G.L.; Tortoreto, M.; Mancini, A.; Addis, A.; Di Cesare, E.; Lenzi, A.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J. Hematol. Oncol. 2014, 7, 46. [Google Scholar] [CrossRef]
- Neggers, J.E.; Vercruysse, T.; Jacquemyn, M.; Vanstreels, E.; Baloglu, E.; Shacham, S.; Crochiere, M.; Landesman, Y.; Daelemans, D. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem. Biol. 2015, 22, 107–116. [Google Scholar] [CrossRef]
- Chen, C.I.; Gutierrez, M.; Brown, P.N.; Gabrail, N.; Baz, R.; Reece, D.E.; Savona, M.; Trudel, S.; Siegel, D.S.; Mau-Sorensen, M.; et al. Anti Tumor Activity Of Selinexor (KPT-330), A First-In-Class Oral Selective Inhibitor Of Nuclear Export (SINE) XPO1/CRM1 Antagonist In Patients (pts) With Relapsed/Refractory Multiple Myeloma (MM) Or Waldenstrom’s Macroglobulinemia (WM). Blood 2013, 122, 1942. [Google Scholar]
- Bergamaschi, A.; Madak-Erdogan, Z.; Kim, Y.J.; Choi, Y.L.; Lu, H.; Katzenellenbogen, B.S. The forkhead transcription factor FOXM1 promotes endocrine resistance and invasiveness in estrogen receptor-positive breast cancer by expansion of stem-like cancer cells. Breast Cancer Res. 2014, 16, 436. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Zhang, X.; Mu, X.; Huang, O.; Xie, Z.; Jiang, M.; Geng, M.; Shen, K. Luminal breast cancer cell lines overexpressing ZNF703 are resistant to tamoxifen through activation of Akt/mTOR signaling. PLoS ONE 2013, 8, e72053. [Google Scholar] [CrossRef]
- Chen, K.L.A.; Zhao, Y.C.; Hieronymi, K.; Smith, B.P.; Madak-Erdogan, Z. Bazedoxifene and conjugated estrogen combination maintains metabolic homeostasis and benefits liver health. PLoS ONE 2017, 12, e0189911. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci. Signal. 2016, 9, ra53. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Madak Erdogan, Z. Systems Biology of Metabolic Regulation by Estrogen Receptor Signaling in Breast Cancer. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Alexander, T.B.; Lacayo, N.J.; Choi, J.K.; Ribeiro, R.C.; Pui, C.-H.; Rubnitz, J.E. Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination with Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. J. Clin. Oncol. 2016, 34, 4094–4101. [Google Scholar] [CrossRef]
- Gounder, M.M.; Zer, A.; Tap, W.D.; Salah, S.; Dickson, M.A.; Gupta, A.A.; Keohan, M.L.; Loong, H.H.; D’Angelo, S.P.; Baker, S.; et al. Phase IB Study of Selinexor, a First-in-Class Inhibitor of Nuclear Export, in Patients With Advanced Refractory Bone or Soft Tissue Sarcoma. J. Clin. Oncol. 2016, 34, 3166–3174. [Google Scholar] [CrossRef]
- Razak, A.R.A.; Mau-Soerensen, M.; Gabrail, N.Y.; Gerecitano, J.F.; Shields, A.F.; Unger, T.J.; Saint-Martin, J.R.; Carlson, R.; Landesman, Y.; McCauley, D.; et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4142–4150. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267. [Google Scholar] [CrossRef]
- Arpino, G.; Gutierrez, C.; Weiss, H.; Rimawi, M.; Massarweh, S.; Bharwani, L.; De Placido, S.; Osborne, C.K.; Schiff, R. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J. Natl. Cancer Inst. 2007, 99, 694–705. [Google Scholar] [CrossRef]
- Wang, C.X.; Koay, D.C.; Edwards, A.; Lu, Z.; Mor, G.; Ocal, I.T.; Digiovanna, M.P. In vitro and in vivo effects of combination of Trastuzumab (Herceptin) and Tamoxifen in breast cancer. Breast Cancer Res. Treat. 2005, 92, 251–263. [Google Scholar] [CrossRef]
- Kondov, B.; Milenkovikj, Z.; Kondov, G.; Petrushevska, G.; Basheska, N.; Bogdanovska-Todorovska, M.; Tolevska, N.; Ivkovski, L. Presentation of the Molecular Subtypes of Breast Cancer Detected By Immunohistochemistry in Surgically Treated Patients. Open Access Maced. J. Med. Sci. 2018, 6, 961–967. [Google Scholar] [CrossRef]
- Inic, Z.; Zegarac, M.; Inic, M.; Markovic, I.; Kozomara, Z.; Djurisic, I.; Inic, I.; Pupic, G.; Jancic, S. Difference between Luminal A and Luminal B Subtypes According to Ki-67, Tumor Size, and Progesterone Receptor Negativity Providing Prognostic Information. Clinical Medicine Insights. Oncology 2014, 8, 107–111. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Charn, T.H.; Jiang, Y.; Liu, E.T.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol. Syst. Biol. 2013, 9, 676. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Hutchinson, K.E.; Stricker, T.P.; Formisano, L.; Young, C.D.; Estrada, M.V.; Nixon, M.J.; Du, L.; Sanchez, V.; Ericsson, P.G.; et al. Genomic profiling of ER+ breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med. 2017, 9, eaai7993. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Jansen, V.M.; Bauer, J.A.; Hanker, A.B.; Sanders, M.E.; González-Ericsson, P.; Kim, S.; Arnedos, M.; André, F.; et al. Abstract 1008: Gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2017, 77, 1008. [Google Scholar] [CrossRef]
- Ranganathan, P.; Kashyap, T.; Yu, X.; Meng, X.; Lai, T.-H.; McNeil, B.; Bhatnagar, B.; Shacham, S.; Kauffman, M.; Dorrance, A.M.; et al. XPO1 Inhibition using Selinexor Synergizes with Chemotherapy in Acute Myeloid Leukemia by Targeting DNA Repair and Restoring Topoisomerase IIα to the Nucleus. Clin. Cancer Res. 2016, 22, 6142–6152. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef]
- Salas Fragomeni, R.A.; Chung, H.W.; Landesman, Y.; Senapedis, W.; Saint-Martin, J.R.; Tsao, H.; Flaherty, K.T.; Shacham, S.; Kauffman, M.; Cusack, J.C. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol. Cancer Ther. 2013, 12, 1171–1179. [Google Scholar] [CrossRef]
- Turner, J.G.; Marchion, D.C.; Dawson, J.L.; Emmons, M.F.; Hazlehurst, L.A.; Washausen, P.; Sullivan, D.M. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009, 69, 6899–6905. [Google Scholar] [CrossRef]
- Hanahan, D.; Robert Weinberg, A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Yndestad, S.; Austreid, E.; Svanberg, I.R.; Knappskog, S.; Lønning, P.E.; Eikesdal, H.P. Activation of Akt characterizes estrogen receptor positive human breast cancers which respond to anthracyclines. Oncotarget 2017, 8, 41227–41241. [Google Scholar] [CrossRef]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef]
- Bostner, J.; Karlsson, E.; Pandiyan, M.J.; Westman, H.; Skoog, L.; Fornander, T.; Nordenskjöld, B.; Stål, O. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res. Treat. 2013, 137, 397–406. [Google Scholar] [CrossRef]
- Zhou, K.; Sun, P.; Zhang, Y.; You, X.; Li, P.; Wang, T. Estrogen stimulated migration and invasion of estrogen receptor-negative breast cancer cells involves an ezrin-dependent crosstalk between G protein-coupled receptor 30 and estrogen receptor beta signaling. Steroids 2016, 111, 113–120. [Google Scholar] [CrossRef]
- Zhang, G. Upregulation of estrogen receptor mediates migration, invasion and proliferation of endometrial carcinoma�cells by regulating the PI3K/AKT/mTOR pathway. Oncol. Rep. 2014, 31, 1175–1182. [Google Scholar]
- Alayev, A.; Salamon, R.S.; Berger, S.M.; Schwartz, N.S.; Cuesta, R.; Snyder, R.B.; Holz, M.K. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene 2016, 35, 3535–3543. [Google Scholar] [CrossRef]
- Shrivastav, A.; Bruce, M.; Jaksic, D.; Bader, T.; Seekallu, S.; Penner, C.; Nugent, Z.; Watson, P.; Murphy, L. The mechanistic target for rapamycin pathway is related to the phosphorylation score for estrogen receptor-alpha in human breast tumors in vivo. Breast Cancer Res. 2014, 16, R49. [Google Scholar] [CrossRef]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 2009, 19, 25–31. [Google Scholar] [CrossRef]
- Michell, B.J.; Griffiths, J.E.; Mitchelhill, K.I.; Rodriguez-Crespo, I.; Tiganis, T.; Bozinovski, S.; de Montellano, P.R.O.; Kemp, B.E.; Pearson, R.B. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 1999, 9, 845–848. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.-P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Wang, Z. Akt Binds to and Phosphorylates Phospholipase C-γ1 in Response to Epidermal Growth Factor. Mol. Biol. Cell 2006, 17, 2267–2277. [Google Scholar] [CrossRef]
- Balz, L.M.; Bartkowiak, K.; Andreas, A.; Pantel, K.; Niggemann, B.; Zänker, K.S.; Brandt, B.H.; Dittmar, T. The interplay of HER2/HER3/PI3K and EGFR/HER2/PLC-γ1 signalling in breast cancer cell migration and dissemination. J. Pathol. 2012, 227, 234–244. [Google Scholar] [CrossRef]
- Ci, Y.; Shi, K.; An, J.; Yang, Y.; Hui, K.; Wu, P.; Shi, L.; Xu, C. ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis. 2014, 5, e1542. [Google Scholar] [CrossRef]
- Liu, B.; Cheng, Y.; Zhang, B.; Bian, H.-J.; Bao, J.-K. Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS–p38–p53 pathway. Cancer Lett. 2009, 275, 54–60. [Google Scholar] [CrossRef]
- Dees, E.C.; Carey, L.A. Improving endocrine therapy for breast cancer: It’s not that simple. J. Clin. Oncol. 2013, 31, 171–173. [Google Scholar] [CrossRef]
- Alluri, P.G.; Speers, C.; Chinnaiyan, A.M. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014, 16, 494. [Google Scholar] [CrossRef]
- Chu, D.; Paoletti, C.; Gersch, C.; Van Den Berg, D.; Zabransky, D.; Cochran, R.; Wong, H.Y.; Valda Toro, P.; Cidado, J.; Croessmann, S.; et al. ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin. Cancer Res. 2016, 22, 993–999. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of Constitutively Active Estrogen Receptor-α Mutations in Pretreated Advanced Estrogen Receptor–Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef]
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968. [Google Scholar] [CrossRef]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci. Transl. Med. 2015, 7, ra182–ra313. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkoyluoglu-Cotul, E.; Smith, B.P.; Wrobel, K.; Chen Zhao, Y.; Chen, K.L.A.; Hieronymi, K.; Imir, O.B.; Duong, K.; O’Callaghan, C.; Mehta, A.; et al. Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance. Cancers 2019, 11, 479. https://doi.org/10.3390/cancers11040479
Kulkoyluoglu-Cotul E, Smith BP, Wrobel K, Chen Zhao Y, Chen KLA, Hieronymi K, Imir OB, Duong K, O’Callaghan C, Mehta A, et al. Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance. Cancers. 2019; 11(4):479. https://doi.org/10.3390/cancers11040479
Chicago/Turabian StyleKulkoyluoglu-Cotul, Eylem, Brandi Patrice Smith, Kinga Wrobel, Yiru Chen Zhao, Karen Lee Ann Chen, Kadriye Hieronymi, Ozan Berk Imir, Kevin Duong, Caitlin O’Callaghan, Aditi Mehta, and et al. 2019. "Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance" Cancers 11, no. 4: 479. https://doi.org/10.3390/cancers11040479