STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment

 , and
, and
Abstract
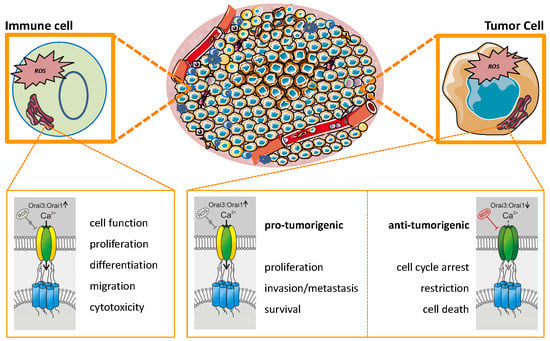
:
1. Introduction
2. The Tumor Microenvironment (TME)
3. STIM/Orai Channels in Cancer
3.1. Short Introduction to STIM and Orai
3.2. STIM/Orai in Tumor Initiation and Promotion
3.3. STIM/Orai in Tumor Proliferation/Growth
3.4. STIM/Orai in Tumor Survival/Apoptosis
3.5. STIM/Orai in Epithelial-to-Mesenchymal Transition (EMT)/Cancer Progression
3.6. STIM/Orai in Tumor Metastasis/Angiogenesis
4. ROS Production and Elimination
5. Impact of Reactive Oxygen Species (ROS) in the Tumor Microenvironment (TME)
5.1. How Can ROS Support Carcinogenesis?
5.2. Sources of ROS in The Tumor Microenvironment
5.3. What Are The Downstream Effects of Increased ROS in The Tumor Microenvironment?
6. Impact of ROS on STIM/Orai Channels
6.1. Impact of ROS on Orai
6.2. Impact of ROS on STIM
7. Interactions between TME, Orai, and ROS: Promoting or Inhibiting Tumor Progression
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Califano, A.; Alvarez, M.J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 2017, 17, 116–130. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim. Biophys. Acta 2016, 1863, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Kinet, J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. Biophys. Acta 2013, 1833, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Alansary, D.; Schmidt, B.; Dorr, K.; Bogeski, I.; Rieger, H.; Kless, A.; Niemeyer, B.A. Thiol dependent intramolecular locking of Orai1 channels. Sci. Rep. 2016, 6, 33347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Gibhardt, C.S.; Schmidt, B.; Lis, A.; Pasieka, B.; Conrad, D.; Jung, P.; Gaupp, R.; Wonnenberg, B.; Diler, E.; et al. A calcium-redox feedback loop controls human monocyte immune responses: The role of ORAI Ca2+ channels. Sci. Signal. 2016, 9, ra26. [Google Scholar] [CrossRef]
- Siddiqui, I.A.; Sanna, V.; Ahmad, N.; Sechi, M.; Mukhtar, H. Resveratrol nanoformulation for cancer prevention and therapy. Ann. N. Y. Acad. Sci. 2015, 1348, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Anari, F.; Ramamurthy, C.; Zibelman, M. Impact of tumor microenvironment composition on therapeutic responses and clinical outcomes in cancer. Future Oncol. 2018, 14, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Weber, C.E.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Vogel, B.; Mehta, S.R.; Mehran, R. Reperfusion strategies in acute myocardial infarction and multivessel disease. Nat. Rev. Cardiol. 2017, 14, 665–678. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Subedi, K.P.; Ong, H.L.; Son, G.Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Bird, G.S.; Hwang, S.Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W., Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009, 19, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Putney, J.W., Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J. Biol. Chem. 2007, 282, 17548–17556. [Google Scholar] [CrossRef] [PubMed]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef]
- Yen, T.T.; Yang, A.; Chiu, W.T.; Li, T.N.; Wang, L.H.; Wu, Y.H.; Wang, H.C.; Chen, L.; Wang, W.C.; Huang, W.; et al. Hepatitis B virus PreS2-mutant large surface antigen activates store-operated calcium entry and promotes chromosome instability. Oncotarget 2016, 7, 23346–23360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Wang, Z.; Han, J.; Ma, H.; Li, Z. High Expression of Orai1 Enhances Cell Proliferation and is Associated with Poor Prognosis in Human Colorectal Cancer. Clin. Lab. 2016, 62, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.Y.; Zhong, L.X.; Feng, M.; Wang, J.F.; Liu, D.B.; Xiong, J.P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef] [Green Version]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef]
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrimgeour, N.; Litjens, T.; Ma, L.; Barritt, G.J.; Rychkov, G.Y. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J. Physiol. 2009, 587, 2903–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, P.J.; Lewis, R.S. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, L.; Deng, Y.; Ji, W.; Du, W.; Xu, P.; Chen, L.; Xu, T. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011, 21, 305–315. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Wang, J.Y.; Sun, J.; Huang, M.Y.; Wang, Y.S.; Hou, M.F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.J.; Lin, S.; et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, P.; Li, J.; Li, F.; Zhu, J.; Gao, P.; Zang, Y.; Wang, Y.; Shan, Y.; Yang, D. Suppression of STIM1 inhibits the migration and invasion of human prostate cancer cells and is associated with PI3K/Akt signaling inactivation. Oncol. Rep. 2017, 38, 2629–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, Z.; Wang, R.; Ma, W.; Yang, Y.; Wei, J.; Wei, Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J. Exp. Clin. Cancer Res. 2013, 32, 20. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Bian, X.Y.; Chen, Q.; Yao, X.F.; Wang, X.D.; Zhang, W.C.; Tao, Y.J.; Jin, R.; Zhang, L. Blocking of stromal interaction molecule 1 expression influence cell proliferation and promote cell apoptosis in vitro and inhibit tumor growth in vivo in head and neck squamous cell carcinoma. PLoS ONE 2017, 12, e0177484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, J.; Jiang, H.; Zhang, Q.; Yang, H.; Xu, X.; Zhang, C.; Xu, C.; Wang, J.; Lu, W. Nicotine enhances storeoperated calcium entry by upregulating HIF1alpha and SOCC components in nonsmall cell lung cancer cells. Oncol. Rep. 2018, 40, 2097–2104. [Google Scholar] [PubMed]
- Choi, S.; Cui, C.; Luo, Y.; Kim, S.H.; Ko, J.K.; Huo, X.; Ma, J.; Fu, L.W.; Souza, R.F.; Korichneva, I.; et al. Selective inhibitory effects of zinc on cell proliferation in esophageal squamous cell carcinoma through Orai1. FASEB J. 2018, 32, 404–416. [Google Scholar] [CrossRef]
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Mejean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 Prevents Calcium-Induced Progression of Early-Stage Prostate Tumors by Counteracting TRPC6 and Calcium Sensing Receptor Upregulation. Cancer Res. 2017, 77, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Song, W.; Li, L.; Cao, L.; Yu, Y.; Song, C.; Wang, Y.; Zhang, F.; Li, Y.; Zhang, B.; et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget 2016, 7, 74015–74030. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Liu, J.; Sun, Q.; Chen, R.; Wang, Y.; Duan, J.; Li, C.; Li, B.; Jing, Y.; Chen, X.; et al. mTORC1 enhancement of STIM1-mediated store-operated Ca2+ entry constrains tuberous sclerosis complex-related tumor development. Oncogene 2013, 32, 4702–4711. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [PubMed]
- Aytes, A.; Mollevi, D.G.; Martinez-Iniesta, M.; Nadal, M.; Vidal, A.; Morales, A.; Salazar, R.; Capella, G.; Villanueva, A. Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype. Mol. Carcinog. 2012, 51, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Stanisz, H.; Saul, S.; Muller, C.S.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Ay, A.S.; Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef]
- Vashisht, A.; Tanwar, J.; Motiani, R.K. Regulation of proto-oncogene Orai3 by miR18a/b and miR34a. Cell Calcium 2018, 75, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, M.; Xu, L.; Lin, D.; Cai, S.; Zou, F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp. Toxicol. Pathol. 2013, 65, 1073–1081. [Google Scholar] [CrossRef]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.T.; Chang, H.A.; Lin, Y.H.; Lin, Y.S.; Chang, H.T.; Lin, H.H.; Huang, S.C.; Tang, M.J.; Shen, M.R. Bcl(-)2 regulates store-operated Ca(2+) entry to modulate ER stress-induced apoptosis. Cell Death Discov. 2018, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Li, W.; Zhang, H.; Zha, L.; Xue, Y.; Wu, X.; Zou, F. Requirement for store-operated calcium entry in sodium butyrate-induced apoptosis in human colon cancer cells. Biosci. Rep. 2012, 32, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gueder, N.; Allan, G.; Telliez, M.S.; Hague, F.; Fernandez, J.M.; Sanchez-Fernandez, E.M.; Ortiz-Mellet, C.; Ahidouch, A.; Ouadid-Ahidouch, H. sp(2) -Iminosugar alpha-glucosidase inhibitor 1-C-octyl-2-oxa-3-oxocastanospermine specifically affected breast cancer cell migration through Stim1, beta1-integrin, and FAK signaling pathways. J. Cell. Physiol. 2017, 232, 3631–3640. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, S.; Niu, H.; Ye, Y.; Hu, F.; Chen, S.; Li, X.; Luo, X.; Jiang, S.; Liu, Y.; et al. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial-to-mesenchymal transition in prostate cancer. Sci. Rep. 2015, 5, 11754. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Qin, K.; Zhang, Y.; Gong, J.; Li, N.; Lv, D.; Xiang, R.; Tan, X. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 411, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca(2+) entry and promote TGF-beta-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.B.; Dhasarathy, A. The calcium channel proteins ORAI3 and STIM1 mediate TGF-beta induced Snai1 expression. Oncotarget 2018, 9, 29468–29483. [Google Scholar] [CrossRef] [PubMed]
- Brundage, R.A.; Fogarty, K.E.; Tuft, R.A.; Fay, F.S. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 1991, 254, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ishihara, A.; Oxford, G.; Johnson, B.; Jacobson, K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999, 400, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Mo, P.; Yang, S. The store-operated calcium channels in cancer metastasis: From cell migration, invasion to metastatic colonization. Front. Biosci. (Landmark. Ed.) 2018, 23, 1241–1256. [Google Scholar] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca(2+) oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell. Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef]
- Kadir, S.; Astin, J.W.; Tahtamouni, L.; Martin, P.; Nobes, C.D. Microtubule remodelling is required for the front-rear polarity switch during contact inhibition of locomotion. J. Cell Sci. 2011, 124, 2642–2653. [Google Scholar] [CrossRef]
- Webb, D.J.; Parsons, J.T.; Horwitz, A.F. Adhesion assembly, disassembly and turnover in migrating cells—Over and over and over again. Nat. Cell Biol. 2002, 4, E97–E100. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, Y.F.; Chiu, W.T.; Wang, Y.K.; Chang, H.C.; Shen, M.R. The ER Ca(2)(+) sensor STIM1 regulates actomyosin contractility of migratory cells. J. Cell Sci. 2013, 126, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Chen, Y.F.; Chiu, W.T.; Chen, H.C.; Shen, M.R. STIM1-dependent Ca(2+) signaling regulates podosome formation to facilitate cancer cell invasion. Sci. Rep. 2017, 7, 11523. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Chen, L.; Zhao, P.; Zhou, H.; Zhang, C.; Yu, S.; Lin, Y.; Yang, X. Store-operated Ca(2+) entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J. Exp. Clin. Cancer Res. 2014, 33, 98. [Google Scholar] [CrossRef]
- Huang, C.Y.; Wei, P.L.; Chen, W.Y.; Chang, W.C.; Chang, Y.J. Silencing Heat Shock Protein 27 Inhibits the Progression and Metastasis of Colorectal Cancer (CRC) by Maintaining the Stability of Stromal Interaction Molecule 1 (STIM1) Proteins. Cells 2018, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Emeriau, N.; de Clippele, M.; Gailly, P.; Tajeddine, N. Store operated calcium entry is altered by the inhibition of receptors tyrosine kinase. Oncotarget 2018, 9, 16059–16073. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Hewitson, T.D. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 553–562. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Giaccia, A.J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wan, X.; Kan, H.; Wang, Y.; Yu, F.; Feng, L.; Jin, J.; Zhang, P.; Ma, X. Hypoxia-induced upregulation of Orai1 drives colon cancer invasiveness and angiogenesis. Eur. J. Pharmacol. 2018, 832, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, T.; Wang, Y.; Chen, Z.; Hua, D.; Yao, X.; Ma, X.; Zhang, P. Orai1 is critical for Notch-driven aggressiveness under hypoxic conditions in triple-negative breast cancers. Biochim. Biophys. Acta Mol. Basis Dis 2018, 1864, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J 2006, 25, 129–138. [Google Scholar] [CrossRef]
- Eylenstein, A.; Schmidt, S.; Gu, S.; Yang, W.; Schmid, E.; Schmidt, E.M.; Alesutan, I.; Szteyn, K.; Regel, I.; Shumilina, E.; et al. Transcription factor NF-kappaB regulates expression of pore-forming Ca2+ channel unit, Orai1, and its activator, STIM1, to control Ca2+ entry and affect cellular functions. J. Biol. Chem. 2012, 287, 2719–2730. [Google Scholar] [CrossRef]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dery, M.A.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Makino, A.; Yuan, J.X. STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm. Circul. 2011, 1, 84–94. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.G.; Chalmers, S.B.; Yapa, K.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers 2019, 11, 208. [Google Scholar] [CrossRef]
- Helfinger, V.; Schroder, K. Redox control in cancer development and progression. Mol. Asp. Med. 2018, 63, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Vander Heiden, M.G.; Thompson, C.B. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell Biol. 2000, 20, 5680–5689. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Li, J.M.; Fan, L.M.; Christie, M.R.; Shah, A.M. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol. Cell Biol. 2005, 25, 2320–2330. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Raimundo, N.; Song, L.; Shutt, T.E.; McKay, S.E.; Cotney, J.; Guan, M.X.; Gilliland, T.C.; Hohuan, D.; Santos-Sacchi, J.; Shadel, G.S. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 2012, 148, 716–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Debowska, K.; Debski, D.; Hardy, M.; Jakubowska, M.; Kalyanaraman, B.; Marcinek, A.; Michalski, R.; Michalowski, B.; Ouari, O.; Sikora, A.; et al. Toward selective detection of reactive oxygen and nitrogen species with the use of fluorogenic probes—Limitations, progress, and perspectives. Pharmacol. Rep. PR 2015, 67, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Pouvreau, S. Genetically encoded reactive oxygen species (ROS) and redox indicators. Biotechnol. J. 2014, 9, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Kalyanaraman, B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free Radic. Biol. Med. 2018, 128, 3–22. [Google Scholar] [CrossRef]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A.; American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.J.; Dick, T.P. Fluorescent protein-based redox probes. Antioxid. Redox Signal. 2010, 13, 621–650. [Google Scholar] [CrossRef]
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A genetically encoded H2O2 probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem. Biol. 2013, 8, 535–542. [Google Scholar] [CrossRef]
- Roma, L.P.; Deponte, M.; Riemer, J.; Morgan, B. Mechanisms and Applications of Redox-Sensitive Green Fluorescent Protein-Based Hydrogen Peroxide Probes. Antioxid. Redox Signal. 2018, 29, 552–568. [Google Scholar] [CrossRef]
- Gibhardt, C.S.; Zimmermann, K.M.; Zhang, X.; Belousov, V.V.; Bogeski, I. Imaging calcium and redox signals using genetically encoded fluorescent indicators. Cell Calcium 2016, 60, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Belousov, V.V. HyPer Family Probes: State of the Art. Antioxid. Redox Signal. 2016, 24, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Setsukinai, K.; Urano, Y.; Kakinuma, K.; Majima, H.J.; Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 2003, 278, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Bikineyeva, A.; Dikalov, S.I. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J. Biomol. Screen. 2013, 18, 498–503. [Google Scholar] [CrossRef]
- Bozem, M.; Knapp, P.; Mirceski, V.; Slowik, E.J.; Bogeski, I.; Kappl, R.; Heinemann, C.; Hoth, M. Electrochemical Quantification of Extracellular Local H2O2 Kinetics Originating from Single Cells. Antioxid. Redox Signal. 2018, 29, 501–517. [Google Scholar] [CrossRef]
- Amatore, C.; Arbault, S.; Guille, M.; Lemaitre, F. Electrochemical monitoring of single cell secretion: Vesicular exocytosis and oxidative stress. Chem. Rev. 2008, 108, 2585–2621. [Google Scholar] [CrossRef]
- Wang, Y.; Noel, J.M.; Velmurugan, J.; Nogala, W.; Mirkin, M.V.; Lu, C.; Guille Collignon, M.; Lemaitre, F.; Amatore, C. Nanoelectrodes for determination of reactive oxygen and nitrogen species inside murine macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, 11534–11539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S.I.; Polienko, Y.F.; Kirilyuk, I. Electron Paramagnetic Resonance Measurements of Reactive Oxygen Species by Cyclic Hydroxylamine Spin Probes. Antioxid. Redox Signal. 2018, 28, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mejiba, S.E.; Zhai, Z.; Della-Vedova, M.C.; Munoz, M.D.; Chatterjee, S.; Towner, R.A.; Hensley, K.; Floyd, R.A.; Mason, R.P.; Ramirez, D.C. Immuno-spin trapping from biochemistry to medicine: Advances, challenges, and pitfalls. Focus on protein-centered radicals. Biochim. Biophys. Acta 2014, 1840, 722–729. [Google Scholar] [CrossRef]
- Mason, R.P. Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol. 2016, 8, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Towner, R.A.; Smith, N. In Vivo and In Situ Detection of Macromolecular Free Radicals Using Immuno-Spin Trapping and Molecular Magnetic Resonance Imaging. Antioxid. Redox Signal. 2018, 28, 1404–1415. [Google Scholar] [CrossRef]
- Jena, N.R. DNA damage by reactive species: Mechanisms, mutation and repair. J. Biosci. 2012, 37, 503–517. [Google Scholar] [CrossRef]
- Rundhaug, J.E.; Fischer, S.M. Molecular mechanisms of mouse skin tumor promotion. Cancers 2010, 2, 436–482. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; McGough, R.; Aswad, B.; Block, J.A.; Terek, R. Hypoxia induces HIF-1alpha and VEGF expression in chondrosarcoma cells and chondrocytes. J. Orthop. Res. 2004, 22, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Meng, Q.; Liu, L.Z.; Rojanasakul, Y.; Wang, X.R.; Jiang, B.H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Ameyar, M.; Wisniewska, M.; Weitzman, J.B. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef]
- Basu, S.; Thorat, R.; Dalal, S.N. MMP7 is required to mediate cell invasion and tumor formation upon Plakophilin3 loss. PLoS ONE 2015, 10, e0123979. [Google Scholar] [CrossRef]
- Ho, B.Y.; Wu, Y.M.; Chang, K.J.; Pan, T.M. Dimerumic acid inhibits SW620 cell invasion by attenuating H(2)O(2)-mediated MMP-7 expression via JNK/C-Jun and ERK/C-Fos activation in an AP-1-dependent manner. Int. J. Biol. Sci. 2011, 7, 869–880. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Wang, Z.; Pu, X.; Zhou, S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol. Appl. Pharmacol. 2011, 254, 86–99. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef]
- Meplan, C.; Richard, M.J.; Hainaut, P. Redox signalling and transition metals in the control of the p53 pathway. Biochem. Pharmacol. 2000, 59, 25–33. [Google Scholar] [CrossRef]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Fitzgerald, A.L.; Osman, A.A.; Xie, T.X.; Patel, A.; Skinner, H.; Sandulache, V.; Myers, J.N. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015, 6, e1678. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Masutani, H.; Arai, R.J.; Yamauchi, A.; Hirota, K.; Sakai, T.; Inamoto, T.; Yamaoka, Y.; Yodoi, J.; Nikaido, T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 1999, 274, 35809–35815. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase promotes proliferation of stellate cells and aggravates liver fibrosis induced by bile duct ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Goto, A.; Kurokawa, E.; Hara, H.; Adachi, T. Cross Talk Mechanism among EMT, ROS, and Histone Acetylation in Phorbol Ester-Treated Human Breast Cancer MCF-7 Cells. Oxid. Med. Cell. Longev. 2016, 2016, 1284372. [Google Scholar] [CrossRef] [PubMed]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the E-cadherin promoter. Gastroenterology 2008, 135, 2128–2140. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; Ueshima, K.; Sakurai, T.; et al. Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis. Dig. Dis. 2013, 31, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Bauer, G. Targeting extracellular ROS signaling of tumor cells. Anticancer Res. 2014, 34, 1467–1482. [Google Scholar]
- Jayavelu, A.K.; Moloney, J.N.; Bohmer, F.D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Segal, A.W.; Shatwell, K.P. The NADPH oxidase of phagocytic leukocytes. Ann. N. Y. Acad. Sci. 1997, 832, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. The respiratory burst oxidase. Curr. Opin. Hematol. 1995, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Bertran, E.; Rodriguez-Hernandez, I.; Herraiz, C.; Cantelli, G.; Fabra, A.; Sanz-Moreno, V.; Fabregat, I. The NADPH oxidase NOX4 represses epithelial to amoeboid transition and efficient tumour dissemination. Oncogene 2017, 36, 3002–3014. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell. Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Foyouzi-Youssefi, R.; Petersson, F.; Lew, D.P.; Krause, K.H.; Nusse, O. Chemoattractant-induced respiratory burst: Increases in cytosolic Ca2+ concentrations are essential and synergize with a kinetically distinct second signal. Biochem. J. 1997, 322 Pt 3, 709–718. [Google Scholar] [CrossRef]
- Holzmann, C.; Kilch, T.; Kappel, S.; Armbruster, A.; Jung, V.; Stockle, M.; Bogeski, I.; Schwarz, E.C.; Peinelt, C. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget 2013, 4, 2096–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzmann, C.; Kilch, T.; Kappel, S.; Dorr, K.; Jung, V.; Stockle, M.; Bogeski, I.; Peinelt, C. Differential Redox Regulation of Ca(2)(+) Signaling and Viability in Normal and Malignant Prostate Cells. Biophys. J. 2015, 109, 1410–1419. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Oh-Hora, M.; Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 2008, 20, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Yang, F.; Xu, J.; Tang, L.; Guan, X. Breast cancer stem cell: The roles and therapeutic implications. Cell. Mol. Life Sci. 2016. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca(2)(+) entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin) 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.C.; Kummerow, C.; Wenning, A.S.; Wagner, K.; Sappok, A.; Waggershauser, K.; Griesemer, D.; Strauss, B.; Wolfs, M.J.; Quintana, A.; et al. Calcium dependence of T cell proliferation following focal stimulation. Eur. J. Immunol. 2007, 37, 2723–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul-Pavicic, A.; Chiang, S.C.; Rensing-Ehl, A.; Jessen, B.; Fauriat, C.; Wood, S.M.; Sjoqvist, S.; Hufnagel, M.; Schulze, I.; Bass, T.; et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc. Natl. Acad. Sci. USA 2011, 108, 3324–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemann, C.; Ammann, S.; Heizmann, M.; Fuchs, S.; Bode, S.F.; Heeg, M.; Fuchs, H.; Lehmberg, K.; Zur Stadt, U.; Roll, C.; et al. Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J. Allergy Clin. Immunol. 2017, 140, 1721–1724. [Google Scholar] [CrossRef] [Green Version]
- Backes, C.S.; Friedmann, K.S.; Mang, S.; Knorck, A.; Hoth, M.; Kummerow, C. Natural killer cells induce distinct modes of cancer cell death: Discrimination, quantification, and modulation of apoptosis, necrosis, and mixed forms. J. Biol. Chem. 2018, 293, 16348–16363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knorck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Beel, J.A.; Lillehei, K.O. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, Y.; Roma, L.P.; Sobotta, M.C.; Rose, A.J.; Diaz, M.B.; Locatelli, G.; Breckwoldt, M.O.; Misgeld, T.; Kerschensteiner, M.; Herzig, S.; et al. Mouse redox histology using genetically encoded probes. Sci. Signal. 2016, 9, rs1. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, Y.G.; Bilan, D.S.; Matlashov, M.E.; Mishina, N.M.; Markvicheva, K.N.; Subach, O.M.; Subach, F.V.; Bogeski, I.; Hoth, M.; Enikolopov, G.; et al. Red fluorescent genetically encoded indicator for intracellular hydrogen peroxide. Nat. Commun. 2014, 5, 5222. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique or Method | Tools and Examples | Specificity | (Potential) Applications |
|---|---|---|---|
| Fluorescence- based assays | Dihydroethidium (DHE) | •O2¯, if used with HPLC | in vitro, extra- and intracellular, cell suspensions |
| Dihydrorhodamine (DHR) | not specific | ||
| 2′,7′-dichlorodihydrofluorescein (DCFH2) | not specific | ||
| Amplex Red, Amplex UltraRed | H2O2 | ||
| Hydroxyphenyl Fluorescein (HPF) | not specific | ||
| Aminophenyl Fluorescein (APF) | not specific | ||
| Genetically encoded fluorescent probes | roGFP2 | EGSH | in vivo and in vitro, intracellular, single cells, tissues, subcellular compartments |
| roGFP2 coupled to glutaredoxins | EGSH | ||
| roGFP2 coupled to peroxidases or peroxiredoxins | H2O2 | ||
| HyPer (different variants including HyPer-Red) | H2O2 | ||
| Chemiluminescence assays | Lucigenin | not specific | in vitro, cell suspensions |
| Luminol | |||
| Isoluminol | |||
| Enzymatic assays | Cytochrome C, Superoxide dismutase (SOD), Horseradish Peroxidase (HRP) | •O2¯ | in vitro, cell suspensions |
| Chemical assays | Prussian Blue, Paraquat (1,1′-Dimethyl-4,4′bipyridium dichloride), FOX (containing xylenol orange) | H2O2, peroxides and others | in vitro, cell suspensions |
| Electrochemical assays | Electrodes of various types and sizes (macro-, mini-, micro- ultramicro- and nanoelectrodes), arrays, chips; additional redox mediators | •O2¯, H2O2 and other ROS | in vitro; single cells, cell suspensions, extra- and intracellular |
| Electron paramagnetic resonance (EPR) spectroscopy using spin probes | DMPO (5, 5-dimethyl-1-pyrroline-N-oxide) | •O2¯, OH• | in vitro and in vivo, cell suspensions, extra- and intracellular |
| DEPMPO [5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide] | •O2¯, OH• | ||
| Horseradish Peroxidase assay (enzymatic using cyclic hydroxylamines) | H2O2 | ||
| in vivo EPR with different functional spin traps and probes | Various ROS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frisch, J.; Angenendt, A.; Hoth, M.; Prates Roma, L.; Lis, A. STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment. Cancers 2019, 11, 457. https://doi.org/10.3390/cancers11040457
Frisch J, Angenendt A, Hoth M, Prates Roma L, Lis A. STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment. Cancers. 2019; 11(4):457. https://doi.org/10.3390/cancers11040457
Chicago/Turabian StyleFrisch, Janina, Adrian Angenendt, Markus Hoth, Leticia Prates Roma, and Annette Lis. 2019. "STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment" Cancers 11, no. 4: 457. https://doi.org/10.3390/cancers11040457