Importance of TRAIL Molecular Anatomy in Receptor Oligomerization and Signaling. Implications for Cancer Therapy

 , and
, and
Abstract
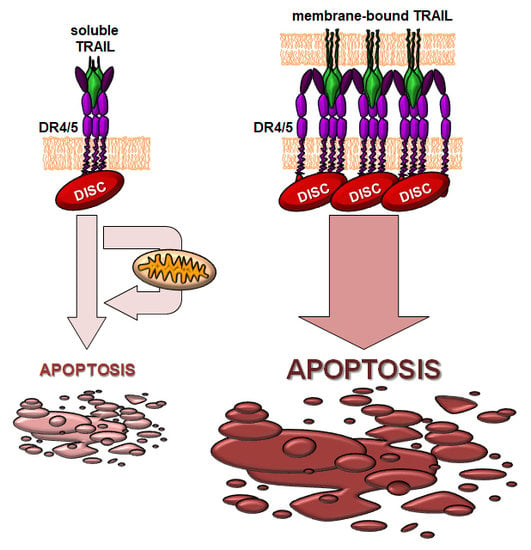
:
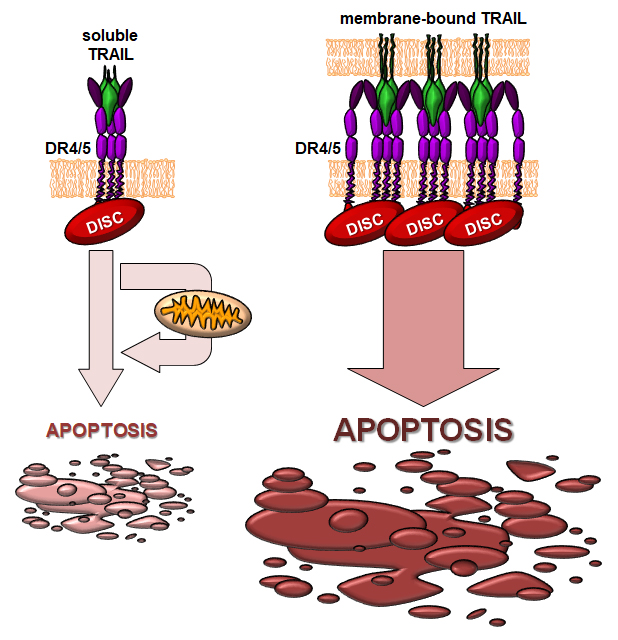{kind=link}
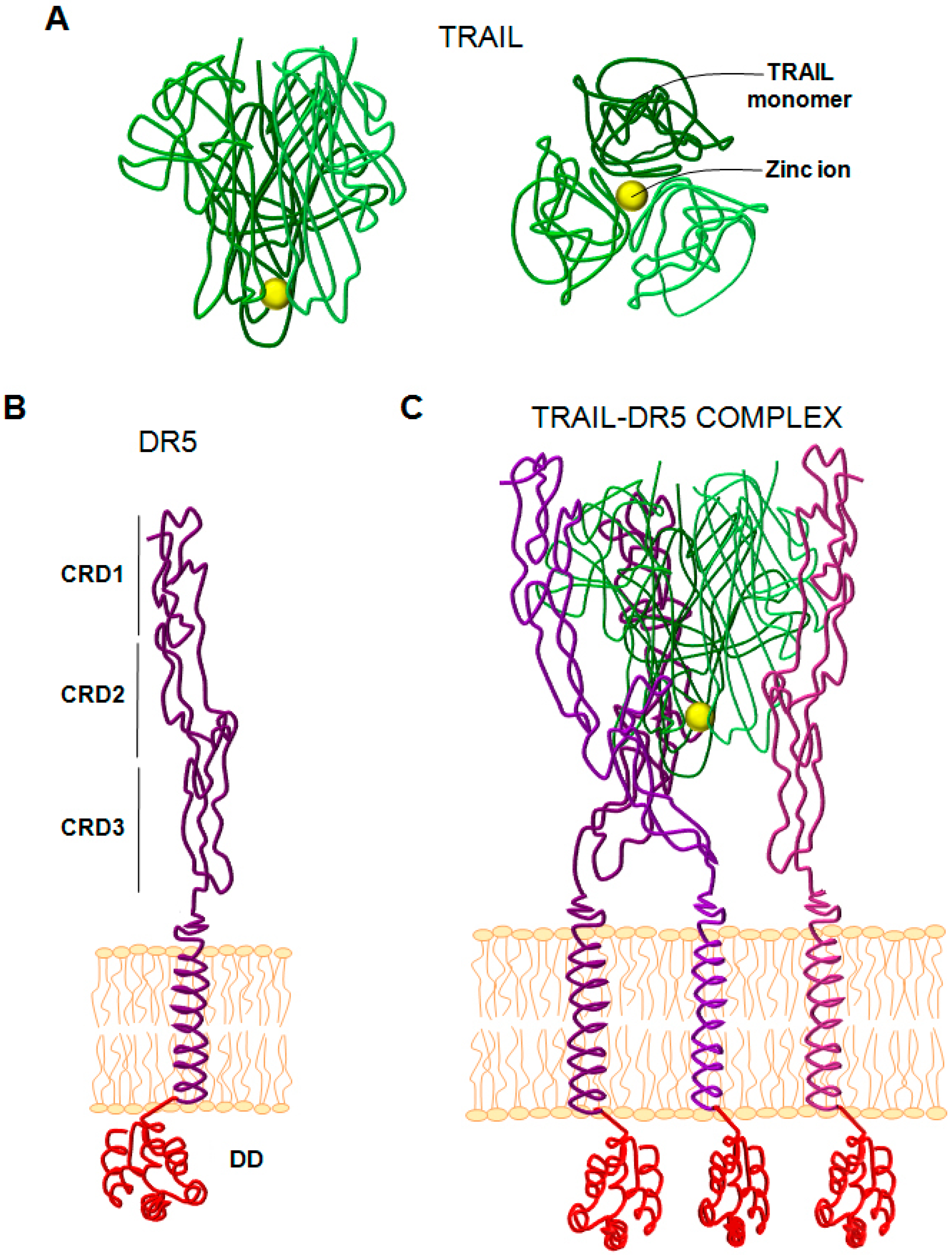{kind=link}
{kind=link}
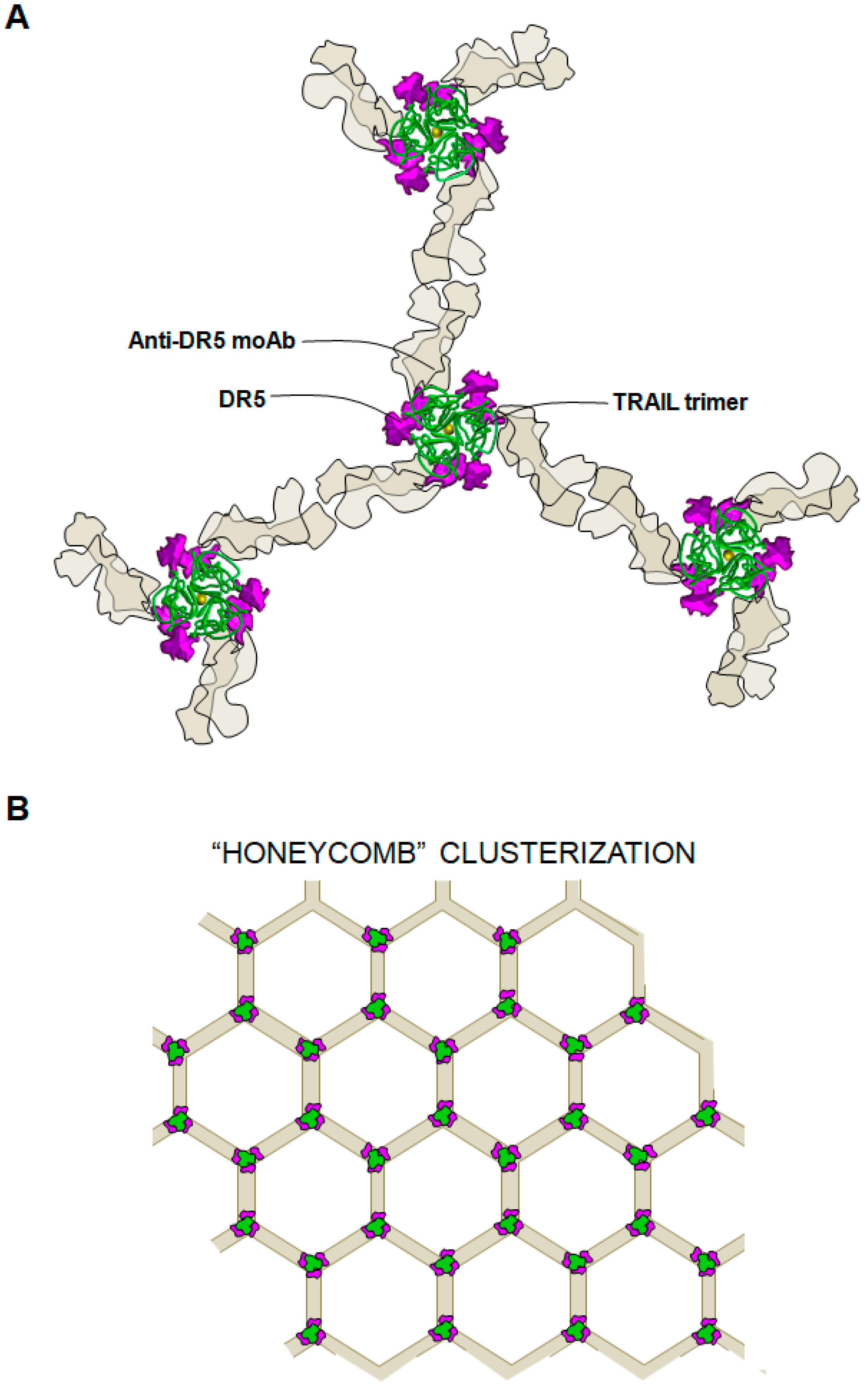{kind=link}
{kind=link}
1. Introduction
2. TRAIL and TRAIL Receptors Structure and Signaling
3. Expression and Secretion of Endogenous TRAIL
4. TRAIL in Cancer Immunosurveillance
5. TRAIL and TRAIL Receptors Oligomerization
6. Implication of TRAIL Receptors Oligomerization in Cancer Therapy
6.1. Recombinant Forms of Human TRAIL
6.1.1. Fusion Proteins
6.1.2. TRAIL-Mimetic Peptides
6.2. TRAIL Receptors Agonists
6.3. TRAIL Anchored to Surfaces
Author Contributions
Funding
Conflicts of Interest
References
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [Green Version]
- Menke, C.; Bin, L.; Thorburn, J.; Behbakht, K.; Ford, H.L.; Thorburn, A. Distinct TRAIL resistance mechanisms can be overcome by proteasome inhibition but not generally by synergizing agents. Cancer Res. 2011, 71, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheah, C.Y.; Belada, D.; Fanale, M.A.; Janikova, A.; Czucman, M.S.; Flinn, I.W.; Kapp, A.V.; Ashkenazi, A.; Kelley, S.; Bray, G.L.; et al. Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: An open-label phase 1b/2 randomised study. Lancet Haematol. 2015, 2, e166–e174. [Google Scholar] [CrossRef]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef]
- Balsas, P.; Lopez-Royuela, N.; Galan-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strekalova, E.; Malin, D.; Rajanala, H.; Cryns, V.L. Metformin sensitizes triple-negative breast cancer to proapoptotic TRAIL receptor agonists by suppressing XIAP expression. Breast Cancer Res. Treat. 2017, 163, 435–447. [Google Scholar] [CrossRef]
- Mauro-Lizcano, M.; Lopez-Rivas, A. Glutamine metabolism regulates FLIP expression and sensitivity to TRAIL in triple-negative breast cancer cells. Cell Death Dis. 2018, 9, 205. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Benito, M.; Martinez-Lorenzo, M.J.; Anel, A.; Marzo, I.; Naval, J. Membrane expression of DR4, DR5 and caspase-8 levels, but not Mcl-1, determine sensitivity of human myeloma cells to Apo2L/TRAIL. Exp. Cell Res. 2007, 313, 2378–2388. [Google Scholar] [CrossRef]
- Hassanzadeh, A.; Farshdousti Hagh, M.; Alivand, M.R.; Akbari, A.A.M.; Shams Asenjan, K.; Saraei, R.; Solali, S. Down-regulation of intracellular anti-apoptotic proteins, particularly c-FLIP by therapeutic agents; the novel view to overcome resistance to TRAIL. J. Cell. Physiol. 2018, 233, 6470–6485. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ji, Y.; Wang, X.; Evers, B.M. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem. Biophys. Res. Commun. 2000, 276, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.S.; Kim, M.S.; Choi, Y.H.; Sung, B.J.; Shin, N.K.; Shin, H.C.; Sung, Y.C.; Oh, B.H. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity 1999, 11, 253–261. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; O’Connell, M.P.; Ultsch, M.H.; Hurst, A.; Totpal, K.; Ashkenazi, A.; de Vos, A.M.; Kelley, R.F. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 2000, 39, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Huang, A.; Skubatch, M.; Baldwin, D.; Yuan, J.; Gurney, A.; Goddard, A.D.; Godowski, P.; et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 1997, 7, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef] [PubMed]
- Morizot, A.; Merino, D.; Lalaoui, N.; Jacquemin, G.; Granci, V.; Iessi, E.; Lanneau, D.; Bouyer, F.; Solary, E.; Chauffert, B.; et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell Death Differ. 2011, 18, 700–711. [Google Scholar] [CrossRef]
- Itoh, N.; Tsujimoto, Y.; Nagata, S. Effect of bcl-2 on Fas antigen-mediated cell death. J. Immunol. 1993, 151, 621–627. [Google Scholar] [PubMed]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74, 845–853. [Google Scholar] [CrossRef]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Hirata, H.; Takahashi, A.; Kobayashi, S.; Yonehara, S.; Sawai, H.; Okazaki, T.; Yamamoto, K.; Sasada, M. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J. Exp. Med. 1998, 187, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K. Three is better than one: Pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine 2007, 37, 101–107. [Google Scholar] [CrossRef]
- Clancy, L.; Mruk, K.; Archer, K.; Woelfel, M.; Mongkolsapaya, J.; Screaton, G.; Lenardo, M.J.; Chan, F.K. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 18099–18104. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000, 288, 2354–2357. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Tardivel, A.; Kovacsovics-Bankowski, M.; Hertig, S.; Gaide, O.; Martinon, F.; Tinel, A.; Deperthes, D.; Calderara, S.; Schulthess, T.; et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 2003, 23, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- O’ Reilly, L.; Tai, L.; Lee, L.; Kruse, E.A.; Grabow, S.; Fairlie, W.D.; Haynes, N.M.; Tarlinton, D.M.; Zhang, J.G.; Belz, G.T.; et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 2009, 461, 659–663. [Google Scholar] [CrossRef]
- Martinez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monle n, I.; Lasierra, P.; Larrad, L.; Pineiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar]
- Schneider, P.; Holler, N.; Bodmer, J.L.; Hahne, M.; Frei, K.; Fontana, A.; Tschopp, J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 1998, 187, 1205–1213. [Google Scholar] [CrossRef]
- Suda, T.; Hashimoto, H.; Tanaka, M.; Ochi, T.; Nagata, S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J. Exp. Med. 1997, 186, 2045–2050. [Google Scholar] [CrossRef]
- Tanaka, M.; Itai, T.; Adachi, M.; Nagata, S. Downregulation of Fas ligand by shedding. Nat. Med. 1998, 4, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Anel, A.; Bosque, A.; Naval, J.; Pineiro, A.; Larrad, L.; Alava, M.A.; Martinez-Lorenzo, M.J. Apo2L/TRAIL and immune regulation. Front. Biosci. 2007, 12, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s path in the immune system. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Marzo, I.; Anel, A.; Naval, J. Targeting the Apo2L/TRAIL system for the therapy of autoimmune diseases and cancer. Biochem. Pharmacol. 2012, 83, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Liu, S.; Yu, Y.; Zhang, M.; Wang, W.; Cao, X. The involvement of TNF-alpha-related apoptosis-inducing ligand in the enhanced cytotoxicity of IFN-beta-stimulated human dendritic cells to tumor cells. J. Immunol. 2001, 166, 5407–5415. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Crafton, E.; Fan, H.N.; Flook, J.; Yoshimura, K.; Skarica, M.; Brockstedt, D.; Dubensky, T.W.; Stins, M.F.; Lanier, L.L.; et al. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat. Med. 2006, 12, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Chaput, N.; Menard, C.; Apetoh, L.; Ullrich, E.; Bonmort, M.; Pequignot, M.; Casares, N.; Terme, M.; Flament, C.; et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nat. Med. 2006, 12, 214–219. [Google Scholar] [CrossRef]
- Dorothee, G.; Vergnon, I.; Menez, J.; Echchakir, H.; Grunenwald, D.; Kubin, M.; Chouaib, S.; Mami-Chouaib, F. Tumor-infiltrating CD4+ T lymphocytes express APO2 ligand (APO2L)/TRAIL upon specific stimulation with autologous lung carcinoma cells: Role of IFN-alpha on APO2L/TRAIL expression and -mediated cytotoxicity. J. Immunol. 2002, 169, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.; Infante-Duarte, C.; Seeger, B.; Zipp, F. Regulation of soluble and surface-bound TRAIL in human T cells, B cells, and monocytes. Cytokine 2003, 24, 244–253. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Monleon, I.; Martinez-Lorenzo, M.J.; Anel, A.; Lasierra, P.; Larrad, L.; Pineiro, A.; Naval, J.; Alava, M.A. CD59 cross-linking induces secretion of APO2 ligand in overactivated human T cells. Eur. J. Immunol. 2000, 30, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Monleon, I.; Martinez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taules, M.; Iturralde, M.; Pineiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J. Immunol. 2001, 167, 6736–6744. [Google Scholar] [CrossRef]
- Schulte, M.; Reiss, K.; Lettau, M.; Maretzky, T.; Ludwig, A.; Hartmann, D.; de Strooper, B.; Janssen, O.; Saftig, P. ADAM10 regulates FasL cell surface expression and modulates FasL-induced cytotoxicity and activation-induced cell death. Cell Death Differ. 2007, 14, 1040–1049. [Google Scholar] [CrossRef] [Green Version]
- Knox, P.G.; Milner, A.E.; Green, N.K.; Eliopoulos, A.G.; Young, L.S. Inhibition of metalloproteinase cleavage enhances the cytotoxicity of Fas ligand. J. Immunol. 2003, 170, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar] [PubMed]
- Secchiero, P.; Gonelli, A.; Corallini, F.; Ceconi, C.; Ferrari, R.; Zauli, G. Metalloproteinase 2 cleaves in vitro recombinant TRAIL: Potential implications for the decreased serum levels of TRAIL after acute myocardial infarction. Atherosclerosis 2010, 211, 333–336. [Google Scholar] [CrossRef]
- Mariani, S.M.; Krammer, P.H. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 1998, 28, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Chaojun, S.; Chunyan, W.; Boquan, J.; Wei, J.; Duo, Z. Metalloprotease inhibitors reducing the shedding of human TRAIL. Proc. 2011 Int. Conf. Hum. Health Biomed. Eng. 2011, 169, 108–111. [Google Scholar] [CrossRef]
- Bosque, A.; Dietz, L.; Gallego-Lleyda, A.; Sanclemente, M.; Iturralde, M.; Naval, J.; Alava, M.A.; Martinez-Lostao, L.; Thierse, H.J.; Anel, A. Comparative proteomics of exosomes secreted by tumoral Jurkat T cells and normal human T cell blasts unravels a potential tumorigenic role for valosin-containing protein. Oncotarget 2016, 7, 29287–29305. [Google Scholar] [CrossRef] [PubMed]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef]
- Seki, N.; Hayakawa, Y.; Brooks, A.D.; Wine, J.; Wiltrout, R.H.; Yagita, H.; Tanner, J.E.; Smyth, M.J.; Sayers, T.J. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis is an important endogenous mechanism for resistance to liver metastases in murine renal cancer. Cancer Res. 2003, 63, 207–213. [Google Scholar] [PubMed]
- Sedger, L.M.; Glaccum, M.B.; Schuh, J.C.; Kanaly, S.T.; Williamson, E.; Kayagaki, N.; Yun, T.; Smolak, P.; Le, T.; Goodwin, R.; et al. Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur. J. Immunol. 2002, 32, 2246–2254. [Google Scholar] [CrossRef]
- Finnberg, N.; Klein-Szanto, A.J.; El-Deiry, W.S. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J. Clin. Investig. 2008, 118, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Diehl, G.E.; Yue, H.H.; Hsieh, K.; Kuang, A.A.; Ho, M.; Morici, L.A.; Lenz, L.L.; Cado, D.; Riley, L.W.; Winoto, A. TRAIL-R as a negative regulator of innate immune cell responses. Immunity 2004, 21, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Zerafa, N.; Westwood, J.A.; Cretney, E.; Mitchell, S.; Waring, P.; Iezzi, M.; Smyth, M.J. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J. Immunol. 2005, 175, 5586–5590. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.H.; Diehl, G.E.; Winoto, A. Loss of TRAIL-R does not affect thymic or intestinal tumor development in p53 and adenomatous polyposis coli mutant mice. Cell Death Differ. 2005, 12, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailey, S.L.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schutz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Investig. 2008, 118, 100–110. [Google Scholar] [CrossRef]
- Von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer Cell-Autonomous TRAIL-R Signaling Promotes KRAS-Driven Cancer Progression, Invasion, and Metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Henkler, F.; Behrle, E.; Dennehy, K.M.; Wicovsky, A.; Peters, N.; Warnke, C.; Pfizenmaier, K.; Wajant, H. The extracellular domains of FasL and Fas are sufficient for the formation of supramolecular FasL-Fas clusters of high stability. J. Cell Biol. 2005, 168, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Berg, D.; Lehne, M.; Muller, N.; Siegmund, D.; Munkel, S.; Sebald, W.; Pfizenmaier, K.; Wajant, H. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ. 2007, 14, 2021–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Yang, C.Y.; Chien, W.L.; Lin, K.I.; Lai, M.Z. Removal of syndecan-1 promotes TRAIL-induced apoptosis in myeloma cells. J. Immunol. 2012, 188, 2914–2921. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Quax, W.J.; Mark, A.E. The conformation of the extracellular binding domain of Death Receptor 5 in the presence and absence of the activating ligand TRAIL: A molecular dynamics study. Proteins 2008, 70, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; van der Sloot, A.M.; Mahalingam, D.; O’Leary, L.; Cool, R.H.; Munoz, I.G.; Montoya, G.; Quax, W.J.; de Jong, S.; Samali, A.; et al. Kinetics in signal transduction pathways involving promiscuous oligomerizing receptors can be determined by receptor specificity: Apoptosis induction by TRAIL. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef]
- Wajant, H.; Moosmayer, D.; Wuest, T.; Bartke, T.; Gerlach, E.; Schonherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [Green Version]
- Wyzgol, A.; Muller, N.; Fick, A.; Munkel, S.; Grigoleit, G.U.; Pfizenmaier, K.; Wajant, H. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J. Immunol. 2009, 183, 1851–1861. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Lohden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Carrington, P.E.; Sandu, C.; Wei, Y.; Hill, J.M.; Morisawa, G.; Huang, T.; Gavathiotis, E.; Werner, M.H. The structure of FADD and its mode of interaction with procaspase-8. Mol. Cell 2006, 22, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Fu, T.M.; Zhao, W.; Zhao, L.; Chen, W.; Qiu, C.; Liu, W.; Liu, Z.; Piai, A.; Fu, Q.; et al. Higher-Order Clustering of the Transmembrane Anchor of DR5 Drives Signaling. Cell 2019, 176, 1477–1489. [Google Scholar] [CrossRef]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Hughes, M.A.; Schilling, R.; Sticht, C.; Tenev, T.; Ploesser, M.; Meier, P.; Sprick, M.R.; MacFarlane, M.; Leverkus, M. Caspase-10 Negatively Regulates Caspase-8-Mediated Cell Death, Switching the Response to CD95L in Favor of NF-κB Activation and Cell Survival. Cell Rep. 2017, 19, 785–797. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Marsal, J.; Gutta, C.; Eisler, S.A.; Peters, N.; Bethea, J.R.; Pfizenmaier, K.; Kontermann, R.E. Novel strategies to mimic transmembrane tumor necrosis factor-dependent activation of tumor necrosis factor receptor 2. Sci. Rep. 2017, 7, 6607. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; Noack, A.; Adam, D.; Tchikov, V.; Bertsch, U.; Roder, C.; Schutze, S.; Wajant, H.; Kalthoff, H.; Trauzold, A. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J. Mol. Med. 2010, 88, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Muhlenbeck, F.; Schneider, P.; Bodmer, J.L.; Schwenzer, R.; Hauser, A.; Schubert, G.; Scheurich, P.; Moosmayer, D.; Tschopp, J.; Wajant, H. The tumor necrosis factor-related apoptosis-inducing ligand receptors TRAIL-R1 and TRAIL-R2 have distinct cross-linking requirements for initiation of apoptosis and are non-redundant in JNK activation. J. Biol. Chem. 2000, 275, 32208–32213. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; MacFarlane, M.; Inoue, S.; Walewska, R.; Majid, A.; Knee, D.; Stover, D.R.; Dyer, M.J.; Cohen, G.M. TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. Br. J. Haematol. 2007, 139, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Marconi, M.; Ascione, B.; Ciarlo, L.; Vona, R.; Garofalo, T.; Sorice, M.; Gianni, A.M.; Locatelli, S.L.; Carlo-Stella, C.; Malorni, W.; et al. Constitutive localization of DR4 in lipid rafts is mandatory for TRAIL-induced apoptosis in B-cell hematologic malignancies. Cell Death Dis. 2013, 4, e863. [Google Scholar] [CrossRef]
- Lee, H.W.; Lee, S.H.; Ryu, Y.W.; Kwon, M.H.; Kim, Y.S. Homomeric and heteromeric interactions of the extracellular domains of death receptors and death decoy receptors. Biochem. Biophys. Res. Commun. 2005, 330, 1205–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smulski, C.R.; Decossas, M.; Chekkat, N.; Beyrath, J.; Willen, L.; Guichard, G.; Lorenzetti, R.; Rizzi, M.; Eibel, H.; Schneider, P.; et al. Hetero-oligomerization between the TNF receptor superfamily members CD40, Fas and TRAILR2 modulate CD40 signalling. Cell Death Dis. 2017, 8, e2601. [Google Scholar] [CrossRef]
- Rauert, H.; Wicovsky, A.; Muller, N.; Siegmund, D.; Spindler, V.; Waschke, J.; Kneitz, C.; Wajant, H. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 2010, 285, 7394–7404. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.F.; Totpal, K.; Lindstrom, S.H.; Mathieu, M.; Billeci, K.; Deforge, L.; Pai, R.; Hymowitz, S.G.; Ashkenazi, A. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J. Biol. Chem. 2005, 280, 2205–2212. [Google Scholar] [CrossRef]
- Herbst, R.S.; Kurzrock, R.; Hong, D.S.; Valdivieso, M.; Hsu, C.P.; Goyal, L.; Juan, G.; Hwang, Y.C.; Wong, S.; Hill, J.S.; et al. A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 5883–5891. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz-Ares, L.; Balint, B.; de Boer, R.H.; van Meerbeeck, J.P.; Wierzbicki, R.; De Souza, P.; Galimi, F.; Haddad, V.; Sabin, T.; Hei, Y.J.; et al. A randomized phase 2 study of paclitaxel and carboplatin with or without conatumumab for first-line treatment of advanced non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Mark, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhasz, E.; Pujol, J.L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.M.; Flores, H.; Gogineni, A.; Marsters, S.; Lawrence, D.A.; Kelley, R.F.; Ngu, H.; Sagolla, M.; Komuves, L.; Bourgon, R.; et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc. Natl. Acad. Sci. USA 2015, 112, 5679–5684. [Google Scholar] [CrossRef] [Green Version]
- Prigozhina, T.B.; Szafer, F.; Aronin, A.; Tzdaka, K.; Amsili, S.; Makdasi, E.; Shani, N.; Dranitzki Elhalel, M. Fn14.TRAIL fusion protein is oligomerized by TWEAK into a superefficient TRAIL analog. Cancer Lett. 2017, 400, 99–109. [Google Scholar] [CrossRef]
- Gieffers, C.; Kluge, M.; Merz, C.; Sykora, J.; Thiemann, M.; Schaal, R.; Fischer, C.; Branschadel, M.; Abhari, B.A.; Hohenberger, P.; et al. APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol. Cancer Ther. 2013, 12, 2735–2747. [Google Scholar] [CrossRef]
- Hutt, M.; Marquardt, L.; Seifert, O.; Siegemund, M.; Muller, I.; Kulms, D.; Pfizenmaier, K.; Kontermann, R.E. Superior Properties of Fc-comprising scTRAIL Fusion Proteins. Mol. Cancer Ther. 2017, 16, 2792–2802. [Google Scholar] [CrossRef]
- Schneider, B.; Munkel, S.; Krippner-Heidenreich, A.; Grunwald, I.; Wels, W.S.; Wajant, H.; Pfizenmaier, K.; Gerspach, J. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis. 2010, 1, e68. [Google Scholar] [CrossRef]
- Siegemund, M.; Pollak, N.; Seifert, O.; Wahl, K.; Hanak, K.; Vogel, A.; Nussler, A.K.; Gottsch, D.; Munkel, S.; Bantel, H.; et al. Superior antitumoral activity of dimerized targeted single-chain TRAIL fusion proteins under retention of tumor selectivity. Cell Death Dis. 2012, 3, e295. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Tung, C.H.; Yang, K.; Weissleder, R.; Breakefield, X.O. Inducible release of TRAIL fusion proteins from a proapoptotic form for tumor therapy. Cancer Res. 2004, 64, 3236–3242. [Google Scholar] [CrossRef]
- Stieglmaier, J.; Bremer, E.; Kellner, C.; Liebig, T.M.; ten Cate, B.; Peipp, M.; Schulze-Koops, H.; Pfeiffer, M.; Buhring, H.J.; Greil, J.; et al. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunol. Immunother. 2008, 57, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Wahl, K.; Siegemund, M.; Lehner, F.; Vondran, F.; Nussler, A.; Langer, F.; Krech, T.; Kontermann, R.; Manns, M.P.; Schulze-Osthoff, K.; et al. Increased apoptosis induction in hepatocellular carcinoma by a novel tumor-targeted TRAIL fusion protein combined with bortezomib. Hepatology 2013, 57, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; He, Y.; Falo, L.D., Jr.; Hui, K.M.; Huang, L. Regression of human mammary adenocarcinoma by systemic administration of a recombinant gene encoding the hFlex-TRAIL fusion protein. Mol. Ther. 2001, 3, 368–374. [Google Scholar] [CrossRef]
- Pavet, V.; Beyrath, J.; Pardin, C.; Morizot, A.; Lechner, M.C.; Briand, J.P.; Wendland, M.; Maison, W.; Fournel, S.; Micheau, O.; et al. Multivalent DR5 peptides activate the TRAIL death pathway and exert tumoricidal activity. Cancer Res. 2010, 70, 1101–1110. [Google Scholar] [CrossRef]
- Lamanna, G.; Smulski, C.R.; Chekkat, N.; Estieu-Gionnet, K.; Guichard, G.; Fournel, S.; Bianco, A. Multimerization of an apoptogenic TRAIL-mimicking peptide by using adamantane-based dendrons. Chemistry 2012, 19, 1762–1768. [Google Scholar] [CrossRef]
- Valldorf, B.; Fittler, H.; Deweid, L.; Ebenig, A.; Dickgiesser, S.; Sellmann, C.; Becker, J.; Zielonka, S.; Empting, M.; Avrutina, O.; et al. An Apoptosis-Inducing Peptidic Heptad That Efficiently Clusters Death Receptor 5. Angew. Chem. Int. Ed. 2016, 55, 5085–5089. [Google Scholar] [CrossRef]
- Chuntharapai, A.; Dodge, K.; Grimmer, K.; Schroeder, K.; Marsters, S.A.; Koeppen, H.; Ashkenazi, A.; Kim, K.J. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J. Immunol. 2001, 166, 4891–4898. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Lefko, P.J.; Graves, J.D.; Zoog, S.J.; Pan, Y.; Wall, J.; Branstetter, D.G.; Moriguchi, J.; Coxon, A.; Huard, J.N.; Xu, R.; et al. Conatumumab, a fully human agonist antibody to death receptor 5, induces apoptosis via caspase activation in multiple tumor types. Cancer Biol. Ther. 2010, 9, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113. [Google Scholar] [CrossRef]
- Haynes, N.M.; Hawkins, E.D.; Li, M.; McLaughlin, N.M.; Hammerling, G.J.; Schwendener, R.; Winoto, A.; Wensky, A.; Yagita, H.; Takeda, K.; et al. CD11c+ dendritic cells and B cells contribute to the tumoricidal activity of anti-DR5 antibody therapy in established tumors. J. Immunol. 2010, 185, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cretney, E.; Hayakawa, Y.; Ota, T.; Akiba, H.; Ogasawara, K.; Yagita, H.; Kinoshita, K.; Okumura, K.; Smyth, M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 2005, 105, 2082–2089. [Google Scholar] [CrossRef] [Green Version]
- Plummer, R.; Attard, G.; Pacey, S.; Li, L.; Razak, A.; Perrett, R.; Barrett, M.; Judson, I.; Kaye, S.; Fox, N.L.; et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin. Cancer Res. 2007, 13, 6187–6194. [Google Scholar] [CrossRef]
- Tamada, T.; Shinmi, D.; Ikeda, M.; Yonezawa, Y.; Kataoka, S.; Kuroki, R.; Mori, E.; Motoki, K. TRAIL-R2 Superoligomerization Induced by Human Monoclonal Agonistic Antibody KMTR2. Sci. Rep. 2015, 5, 17936. [Google Scholar] [CrossRef] [Green Version]
- Graves, J.D.; Kordich, J.J.; Huang, T.H.; Piasecki, J.; Bush, T.L.; Sullivan, T.; Foltz, I.N.; Chang, W.; Douangpanya, H.; Dang, T.; et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell 2014, 26, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, M.H.; Montinaro, A.; Zinngrebe, J.; Prieske, K.; Draber, P.; Prieske, S.; Newsom-Davis, T.; von Karstedt, S.; Graves, J.; Walczak, H. TRAIL-R2-specific antibodies and recombinant TRAIL can synergise to kill cancer cells. Oncogene 2014. [Google Scholar] [CrossRef]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 1999, 189, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Kawasaki, A.; Akiba, H.; Okumura, K.; Yagita, H. Involvement of TNF-related apoptosis-inducing ligand in human CD4+ T cell-mediated cytotoxicity. J. Immunol. 1999, 162, 2639–2647. [Google Scholar] [PubMed]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Takeda, K.; Akiba, H.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Okumura, K.; Yagita, H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999, 163, 1906–1913. [Google Scholar]
- De Miguel, D.; Basanez, G.; Sanchez, D.; Malo, P.G.; Marzo, I.; Larrad, L.; Naval, J.; Pardo, J.; Anel, A.; Martinez-Lostao, L. Liposomes decorated with Apo2L/TRAIL overcome chemoresistance of human hematologic tumor cells. Mol. Pharm. 2013, 10, 893–904. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk. Res. 2015, 39, 657–666. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Erviti-Ardanaz, S.; Pazo-Cid, R.; del Agua, C.; Fernandez, L.J.; Ochoa, I.; Anel, A.; Martinez-Lostao, L. TRAIL-coated lipid-nanoparticles overcome resistance to soluble recombinant TRAIL in non-small cell lung cancer cells. Nanotechnology 2016, 27, 185101. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Pawlak, A.; Conde, B.; Ochoa, I.; Fernandez, L.J.; Anel, A.; Martinez-Lostao, L. Improved Anti-Tumor Activity of Novel Highly Bioactive Liposome-Bound TRAIL in Breast Cancer Cells. Recent Pat. Anticancer Drug Discov. 2016, 11, 197–214. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.M.; Pejenaute-Ochoa, D.; Jarauta, V.; Marzo, I.; Fernandez, L.J.; Ochoa, I.; Conde, B.; Anel, A.; et al. High-order TRAIL oligomer formation in TRAIL-coated lipid nanoparticles enhances DR5 cross-linking and increases antitumour effect against colon cancer. Cancer Lett. 2016, 383, 250–260. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, D.; Gallego-Lleyda, A.; Galan-Malo, P.; Rodriguez-Vigil, C.; Marzo, I.; Anel, A.; Martinez-Lostao, L. Immunotherapy with liposome-bound TRAIL overcome partial protection to soluble TRAIL-induced apoptosis offered by down-regulation of Bim in leukemic cells. Clin. Transl. Oncol. 2015, 17, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Lleyda, A.; De Miguel, D.; Anel, A.; Martinez-Lostao, L. Lipid Nanoparticles Decorated with TNF-Related Aptosis-Inducing Ligand (TRAIL) Are More Cytotoxic than Soluble Recombinant TRAIL in Sarcoma. Int. J. Mol. Sci. 2018, 19, 1449. [Google Scholar] [CrossRef]
- Nair, P.; Lu, M.; Petersen, S.; Ashkenazi, A. Apoptosis initiation through the cell-extrinsic pathway. Methods Enzymol. 2015, 544, 99–128. [Google Scholar] [CrossRef]
- Zakaria, A.B.; Picaud, F.; Rattier, T.; Pudlo, M.; Dufour, F.; Saviot, L.; Chassagnon, R.; Lherminier, J.; Gharbi, T.; Micheau, O.; et al. Nanovectorization of TRAIL with single wall carbon nanotubes enhances tumor cell killing. Nano Lett. 2015, 15, 891–895. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naval, J.; de Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Importance of TRAIL Molecular Anatomy in Receptor Oligomerization and Signaling. Implications for Cancer Therapy. Cancers 2019, 11, 444. https://doi.org/10.3390/cancers11040444
Naval J, de Miguel D, Gallego-Lleyda A, Anel A, Martinez-Lostao L. Importance of TRAIL Molecular Anatomy in Receptor Oligomerization and Signaling. Implications for Cancer Therapy. Cancers. 2019; 11(4):444. https://doi.org/10.3390/cancers11040444
Chicago/Turabian StyleNaval, Javier, Diego de Miguel, Ana Gallego-Lleyda, Alberto Anel, and Luis Martinez-Lostao. 2019. "Importance of TRAIL Molecular Anatomy in Receptor Oligomerization and Signaling. Implications for Cancer Therapy" Cancers 11, no. 4: 444. https://doi.org/10.3390/cancers11040444