Overactive IGF1/Insulin Receptors and NRASQ61R Mutation Drive Mechanisms of Resistance to Pazopanib and Define Rational Combination Strategies to Treat Synovial Sarcoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
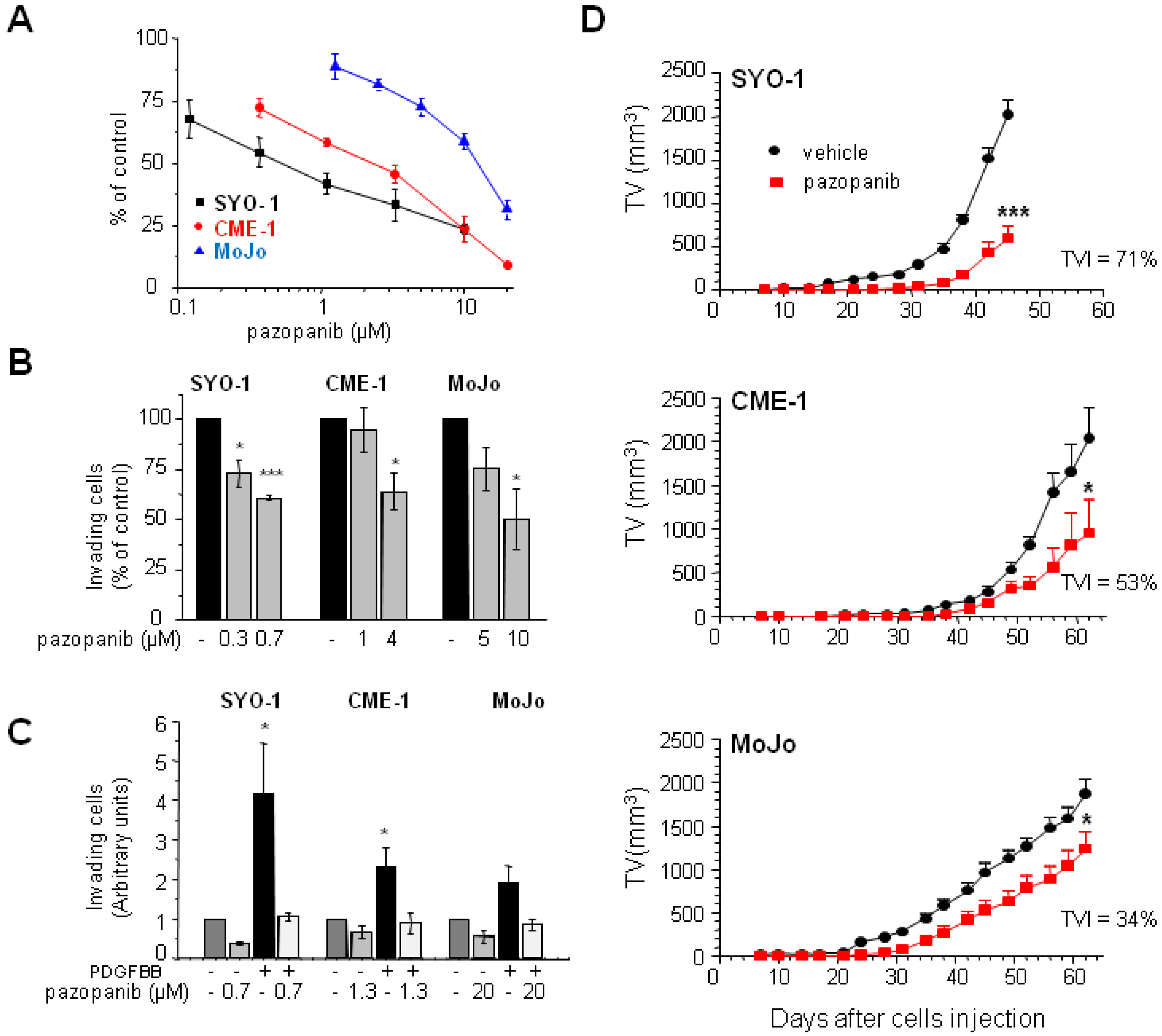
2.1. Human SS Cell Lines Display Different Pazopanib Sensitivity Profiles In Vitro and In Vivo
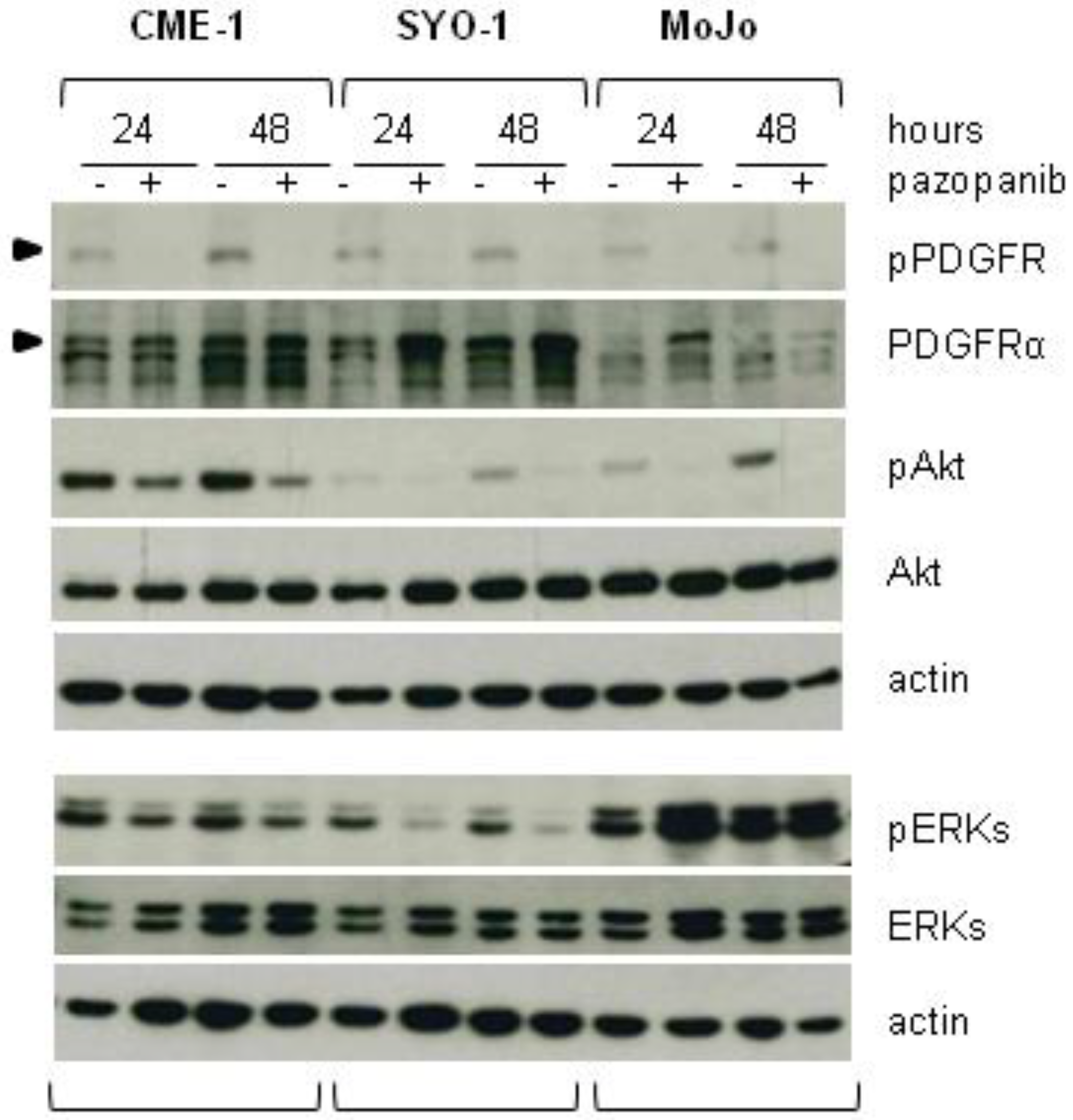
2.2. Reduced Cell Sensitivity to Pazopanib Is Associated with Lowered Inhibition of AKT or ERKs
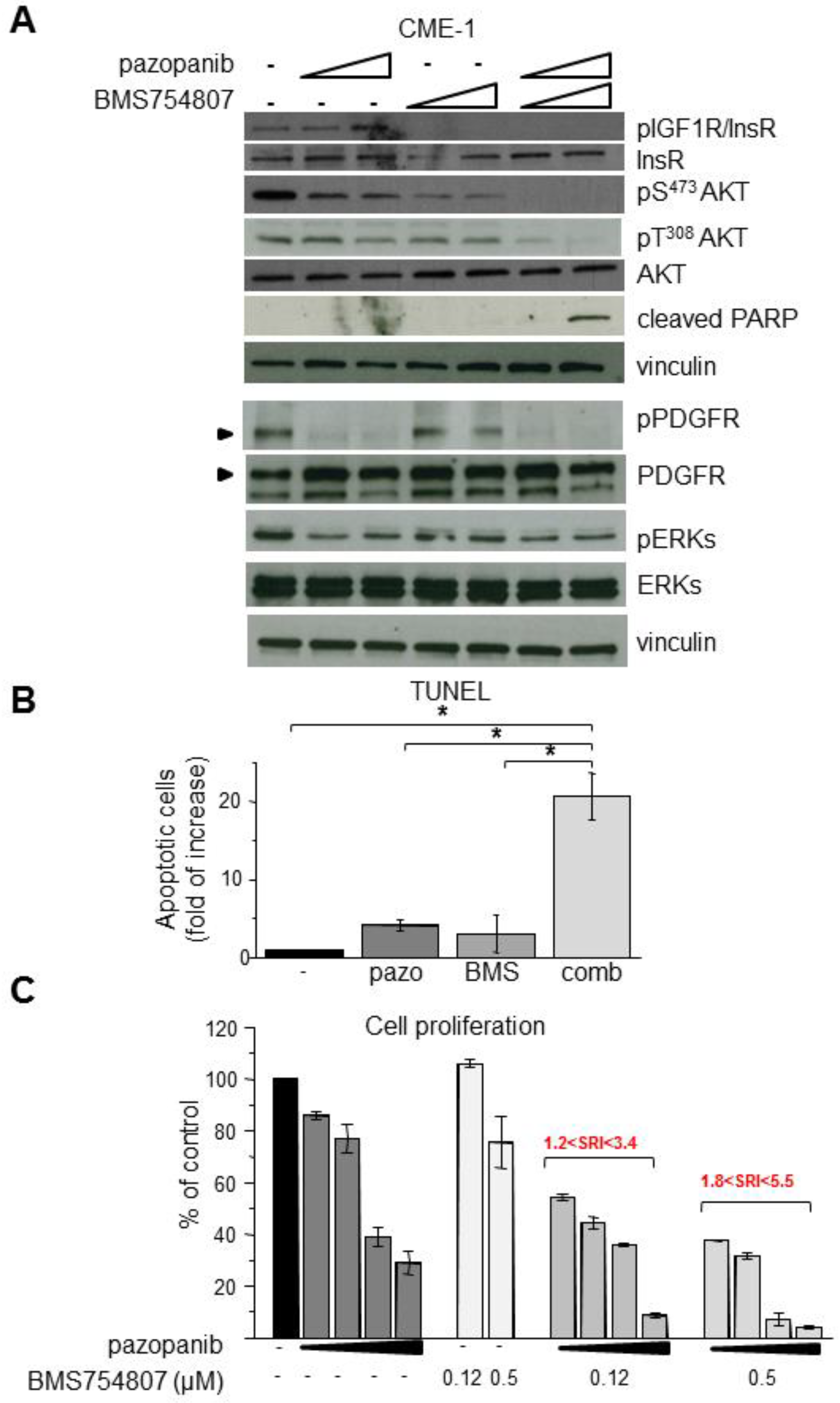
2.3. Inhibition of Overactive IGF1R/InsR Overcomes Pazopanib Resistance in CME-1 Cells
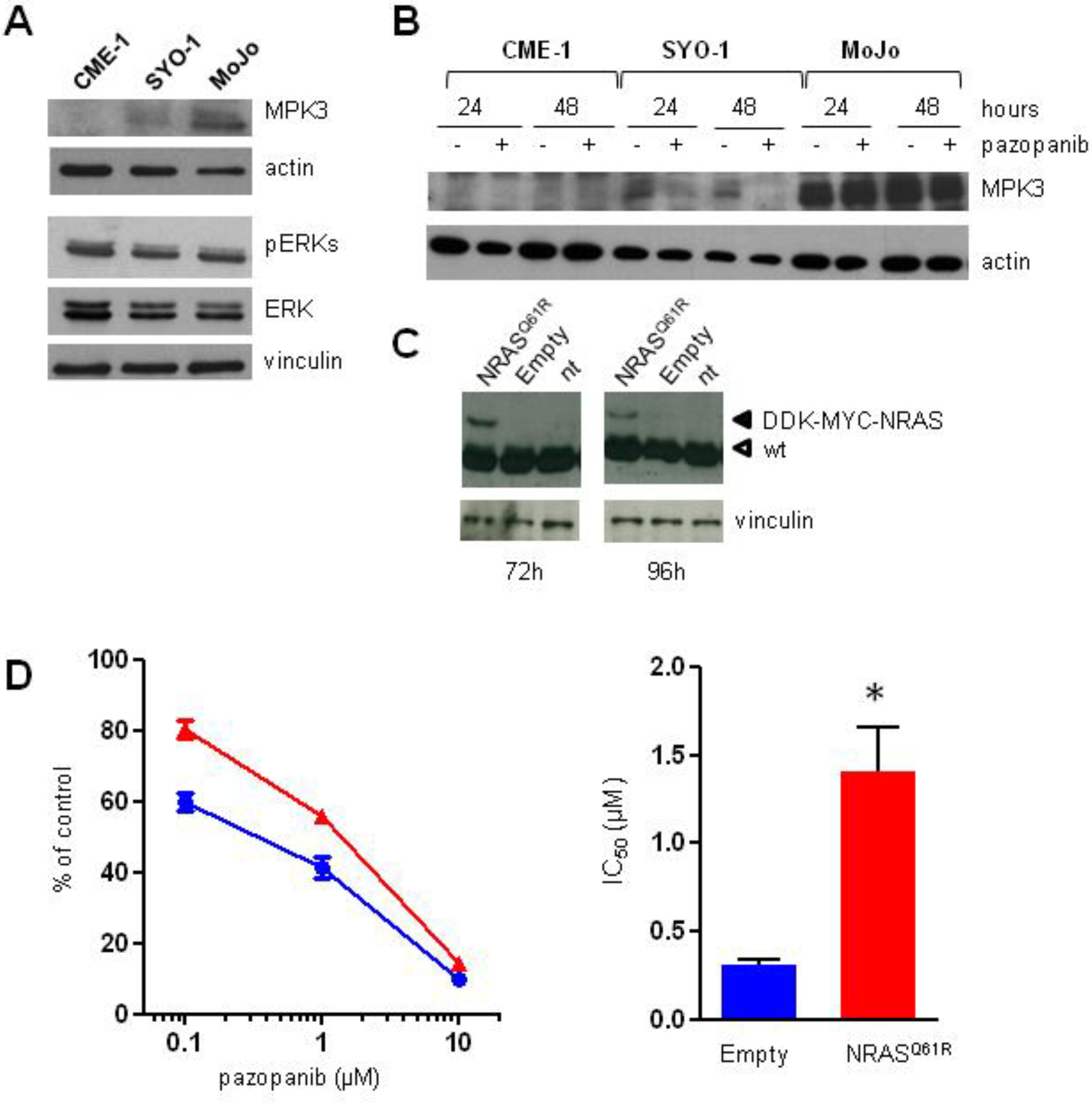
2.4. Aberrant Activation of NRAS/ERKs/MKP3 Pathway in Pazopanib Highly Resistant MoJo Cells
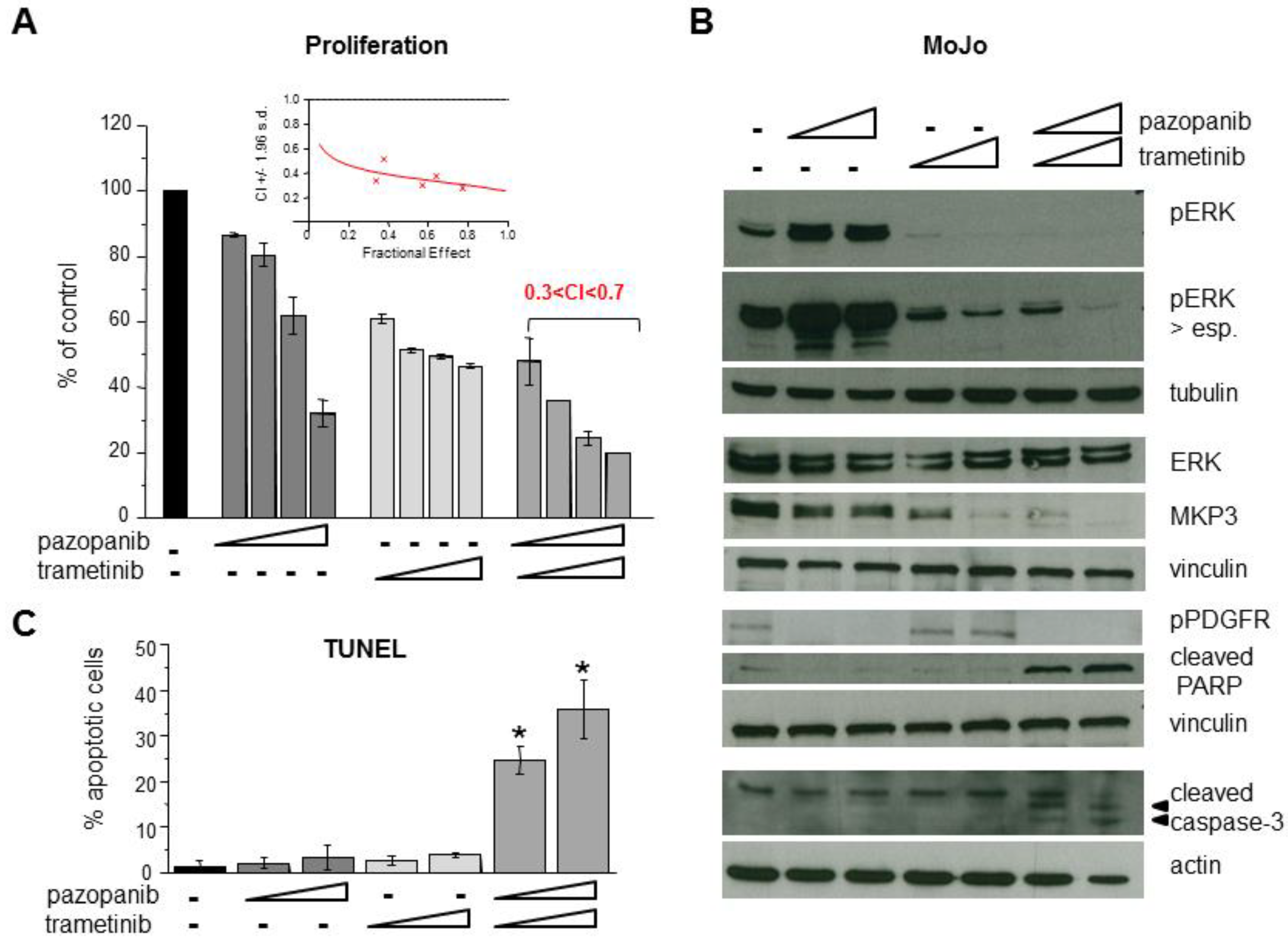
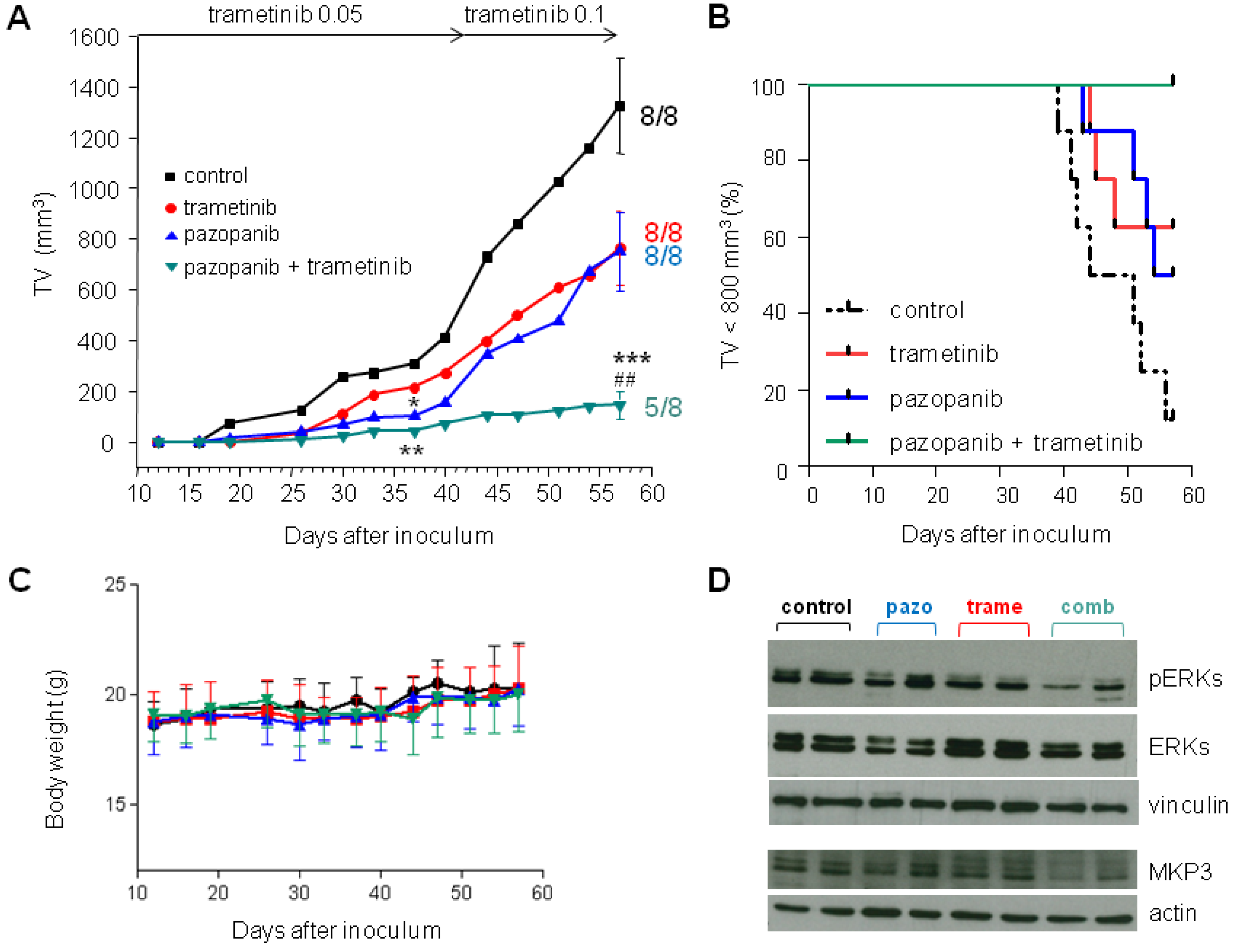
2.5. Inhibition of MEK1/2 Overcomes Pazopanib Resistance in NRASQ61R Mutant MoJo Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Drugs
4.3. Cellular Studies
4.4. Analysis of NRAS Mutational Status
4.5. NRASQ61R Transfection
4.6. Western Blotting
4.7. Antibodies
- Mouse monoclonal: anti-PKBα/Akt from BD Transduction (Lexington, KY, USA); anti-tubulin and anti-vinculin from Sigma; anti-LAMP2 and anti MPK3/DUSP6 from Abcam (Cambridge, UK); anti-pan-Ras from Calbiochem (San Diego, CA, USA); anti-p62 from MBL (Woburn, MA, USA).
- Rabbit monoclonal: anti-phospho-IGF1-IRβ (Tyr1135/1136)/InsRβ (Tyr1150/1151) (19H7) and anti-phospho-PDGFRα (Tyr849)/PDGFRβ (Tyr857) from Cell Signaling Technology (Beverly, MA, USA).
- Rabbit polyclonal: anti-SYT (H-80) and anti-InsRβ (C-19) from Santa Cruz Biotechnology. Anti-cleaved PARP (Asp214), anti-phospho-p44/42 MAPK (Thr202/Tyr204), anti-p44/42 MAPK, anti-phospho-Akt (Ser473), anti-cleaved Caspase-3 (Asp175), and anti-LC3B from Cell Signaling Technology; anti-PDGFRα from Upstate Biotechnology (Lake Placid, NY, USA); anti-pan-Akt phosphoT308 antibody from Abcam; anti-actin from Sigma.
4.8. In Vivo Studies
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sultan, I.; Rodriguez-Galindo, C.; Saab, R.; Yasir, S.; Casanova, M.; Ferrari, A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: An analysis of 1268 patients. Cancer 2009, 115, 3537–3547. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Limon, J.; Gustafson, P.; Skytting, B.; Akerman, M.; Sciot, R.; Dal Cin, P.; Samson, I.; et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer 2001, 31, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A conditional mouse model of synovial sarcoma: Insights into a myogenic origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Carmody Soni, E.E.; Schlottman, S.; Erkizan, H.V.; Uren, A.; Toretsky, J.A. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin. Orthop. Relat. Res. 2014, 472, 874–882. [Google Scholar] [CrossRef]
- Su, L.; Sampaio, A.V.; Jones, K.B.; Pacheco, M.; Goytain, A.; Lin, S.; Poulin, N.; Yi, L.; Rossi, F.M.; Kast, J.; et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: Insights into disease etiology and therapeutics. Cancer Cell 2012, 21, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Stock, R.; Onnasch, D.; Haeckel, C.; Mellin, W.; Franke, D.S.; Roessner, A. Prognostic significance of p53 gene mutations and p53 protein expression in synovial sarcomas. Virchows Arch. 1999, 435, 407–412. [Google Scholar] [CrossRef]
- Oda, Y.; Sakamoto, A.; Satio, T.; Kawauchi, S.; Iwamoto, Y.; Tsuneyoshi, M. Molecular abnormalities of p53, MDM2, and H-ras in synovial sarcoma. Mod. Pathol. 2000, 13, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Vlenterie, M.; Hillebrandt-Roeffen, M.H.; Flucke, U.E.; Groenen, P.J.; Tops, B.B.; Kamping, E.J.; Pfundt, R.; de Bruijn, D.R.; Geurts van Kessel, A.H.; van Krieken, H.J.; et al. Next generation sequencing in synovial sarcoma reveals novel gene mutations. Oncotarget 2015, 6, 34680–34690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desar, I.M.E.; Fleuren, E.D.G.; van der Graaf, W.T.A. Systemic treatment for adults with synovial sarcoma. Curr. Treat. Options Oncol. 2018, 19, 13. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Van Tine, B.A. Synovial Sarcoma: Current Concepts and Future Perspectives. J. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Vlenterie, M.; Litière, S.; Rizzo, E.; Marréaud, S.; Judson, I.; Gelderblom, H.; Le Cesne, A.; Wardelmann, E.; Messiou, C.; Gronchi, A.; et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur. J. Cancer 2016, 58, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Sun, T.; Hou, B.; Hong, G.; Mallampati, S.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; et al. Survival changes in patients with synovial sarcoma, 1983–2012. J. Cancer 2017, 8, 1759–1768. [Google Scholar] [CrossRef]
- Riedel, R.F.; Jones, R.L.; Italiano, A.; Bohac, C.; Thompson, J.C.; Mueller, K.; Khan, Z.; Pollack, S.M.; Van Tine, B.A. Systemic anti-cancer therapy in synovial sarcoma: A systematic review. Cancers 2018, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.L.; Vasudeva, S.D.; Laé, M.; Saito, T.; Barbashina, V.; Antonescu, C.R.; Ladanyi, M.; Schwartz, G.K. PDGF receptor alpha is an alternative mediator of rapamycin-induced Akt activation: Implications for combination targeted therapy of synovial sarcoma. Cancer Res. 2012, 72, 4515–4525. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Vlenterie, M.; van der Graaf, W.T.A.; Hillebrandt-Roeffen, M.H.S.; Blackburn, J.; Ma, X.; Chan, H.; Magias, M.C.; van Erp, A.; van Houdt, L.; et al. Phosphoproteomic profiling reveals ALK and MET as novel actionable targets across synovial sarcoma subtypes. Cancer Res. 2017, 77, 4279–4292. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group, Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Kasper, B.; Sleijfer, S.; Litière, S.; Marreaud, S.; Verweij, J.; Hodge, R.A.; Bauer, S.; Kerst, J.M.; van der Graaf, W.T. Long-term responders and survivors on pazopanib for advanced soft tissue sarcomas: Subanalysis of two European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62043 and 62072. Ann. Oncol. 2014, 25, 719–724. [Google Scholar] [CrossRef]
- Imura, Y.; Nakai, T.; Yamada, S.; Outani, H.; Takenaka, S.; Hamada, K.; Araki, N.; Itoh, K.; Yoshikawa, H.; Naka, N. Functional and therapeutic relevance of hepatocyte growth factor/c-MET signaling in synovial sarcoma. Cancer Sci. 2016, 107, 1867–1876. [Google Scholar] [CrossRef]
- Yokoyama, N.; Matsunobu, T.; Matsumoto, Y.; Fukushi, J.I.; Endo, M.; Hatano, M.; Nabeshima, A.; Fukushima, S.; Okada, S.; Iwamoto, Y. Activation of ERK1/2 causes pazopanib resistance via downregulation of DUSP6 in synovial sarcoma cells. Sci. Rep. 2017, 7, 45332. [Google Scholar] [CrossRef]
- Qiao, Z.; Shiozawa, K.; Kondo, T. Proteomic approach toward determining the molecular background of pazopanib resistance in synovial sarcoma. Oncotarget 2017, 8, 109587–109595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassinelli, G.; Dal Bo, L.; Favini, E.; Cominetti, D.; Pozzi, S.; Tortoreto, M.; De Cesare, M.; Lecis, D.; Scanziani, E.; Minoli, L.; et al. Supersulfated low-molecular weight heparin synergizes with IGF1R/InsR inhibitor to suppress synovial sarcoma growth and metastases. Cancer Lett. 2018, 415, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Ferrari, A.; Negri, T.; Conca, E.; da Luca, R.; Losa, M.; Casieri, P.; Orsenigo, M.; Lampis, A.; Meazza, C.; et al. Molecular characterization of synovial sarcoma in children and adolescents: Evidence of akt activation. Transl. Oncol. 2008, 1, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Pursiheimo, J.P.; Rantanen, K.; Heikkinen, P.T.; Johansen, T.; Jaakkola, P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009, 28, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavallai, S.; Hamed, H.A.; Grant, S.; Poklepovic, A.; Dent, P. Pazopanib and HDAC inhibitors interact to kill sarcoma cells. Cancer Biol. Ther. 2014, 15, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.; Roberts, J.L.; Sander, C.; Lee, J.; Kirkwood, J.M.; Poklepovic, A.; Dent, P. The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 2017, 8, 16367–16386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signaling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kidger, A.M.; Keyse, S.M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 2016, 50, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Vujic, I.; Posch, C.; Sanlorenzo, M.; Yen, A.J.; Tsumura, A.; Kwong, A.; Feichtenschlager, V.; Lai, K.; Arneson, D.V.; Rappersberger, K.; et al. Mutant NRASQ61 shares signaling similarities across various cancer types-potential implications for future therapies. Oncotarget 2014, 5, 7936–7944. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Yoo, K.H.; Kim, H.S.; Lee, S.J.; Park, S.H.; Kim, S.J.; Kim, S.H.; La Choi, Y.; Shin, K.H.; Cho, Y.J.; Lee, J.; et al. Efficacy of pazopanib monotherapy in patients who had been heavily pretreated for metastatic soft tissue sarcoma: A retrospective case series. BMC Cancer 2015, 15, 154. [Google Scholar] [CrossRef] [PubMed]
- Sedef, A.M.; Köse, F.; Doğan, Ö.; Ergün, T.; Sezer, A.; Mertsoylu, H.; Muallaoğlu, S.; Beşen, A.; Özyilkan, Ö. Targeted treatment with pazopanib in metastatic soft tissue sarcoma: Nearly complete response in two cases. Mol. Clin. Oncol. 2015, 3, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.; Basso, E.; Magni, C.; Bergamaschi, L.; Chiaravalli, S.; Carta, R.; Tirtei, E.; Massimino, M.; Fagioli, F.; Ferrari, A. Response to pazopanib in two pediatric patients with pretreated relapsing synovial sarcoma. Tumori J. 2017, 103, e1–e3. [Google Scholar] [CrossRef]
- Friedrichs, N.; Trautmann, M.; Endl, E.; Sievers, E.; Kindler, D.; Wurst, P.; Czerwitzki, J.; Steiner, S.; Renner, M.; Penzel, R.; et al. Phosphatidylinositol-3′-kinase/AKT signaling is essential in synovial sarcoma. Int. J. Cancer 2011, 129, 1564–1575. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Imura, Y.; Nakai, T.; Nakai, S.; Yasuda, N.; Kaneko, K.; Outani, H.; Takenaka, S.; Hamada, K.; Myoui, A.; et al. Therapeutic potential of TAS-115 via c-MET and PDGFRα signal inhibition for synovial sarcoma. BMC Cancer 2017, 17, 334. [Google Scholar] [CrossRef]
- Hosaka, S.; Horiuchi, K.; Yoda, M.; Nakayama, R.; Tohmonda, T.; Susa, M.; Nakamura, M.; Chiba, K.; Toyama, Y.; Morioka, H. A novel multi-kinase inhibitor pazopanib suppresses growth of synovial sarcoma cells through inhibition of the PI3K-AKT pathway. J. Orthop. Res. 2012, 30, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Skytting, B.; Nilsson, G.; Brodin, B.; Larsson, O. Expression of insulin-like growth factor-1 receptor in synovial sarcoma: Association with an aggressive phenotype. Cancer Res. 1999, 59, 3588–3591. [Google Scholar] [PubMed]
- Sun, Y.; Gao, D.; Liu, Y.; Huang, J.; Lessnick, S.; Tanaka, S. IGF2 is critical for tumorigenesis by synovial sarcoma oncoprotein SYT-SSX1. Oncogene 2006, 25, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- De Bruijn, D.R.; Allander, S.V.; van Dijk, A.H.; Willemse, M.P.; Thijssen, J.; van Groningen, J.J.; Meltzer, P.S.; van Kessel, A.G. The synovial-sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regulation. Cancer Res. 2006, 66, 9474–9482. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, N.; Pang, L.J.; Zou, H.; Hu, J.M.; Zhao, J.; Zhang, J.; Liu, C.X.; Zhang, W.J.; Yuan, X.L.; et al. Identification of potential mutations and genomic alterations in the epithelial and spindle cell components of biphasic synovial sarcomas using a human exome SNP chip. BMC Med. Genom. 2015, 8, 69. [Google Scholar] [CrossRef]
- Xing, Z.; Wei, L.; Jiang, X.; Conroy, J.; Glenn, S.; Bshara, W.; Yu, T.; Pao, A.; Tanaka, S.; Kawai, A.; et al. Analysis of mutations in primary and metastatic synovial sarcoma. Oncotarget 2018, 9, 36878–36888. [Google Scholar] [CrossRef]
- Zhang, F.; Cheong, J.K. The renewed battle against RAS-mutant cancers. Cell. Mol. Life Sci. 2016, 73, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.B.; Der, C.J.; Wang-Gillam, A.; Cox, A.D. Targeting RAS-mutant cancers: Is ERK the key? Trends Cancer 2015, 1, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; Greshock, J.; Holbrook, J.D.; Gilmartin, A.; Zhang, X.; McNeil, E.; Conway, T.; Moy, C.; Laquerre, S.; Bachman, K.; et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol. Cancer Ther. 2012, 11, 720–729. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Strudel, M.; Ascierto, P.A. MEK inhibitors in the treatment of metastatic melanoma and solid tumors. Am. J. Clin. Dermatol. 2017, 18, 745–754. [Google Scholar] [CrossRef]
- Khunger, V.; Khunger, M.; Velcheti, V. Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: Clinical evidence and experience. Ther. Adv. Respir. Dis. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Couselo, E.; Adelantado, Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed]
- Ball, D.W.; Jin, N.; Xue, P.; Bhan, S.; Ahmed, S.R.; Rosen, D.M.; Schayowitz, A.; Clark, D.P.; Nelkin, B.D. Trametinib with and without pazopanib has potent preclinical activity in thyroid cancer. Oncol. Rep. 2015, 34, 2319–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, V.L.; Wan, E.; Foo, S.; Nathan, M.R.; Welti, J.C.; Frentzas, S.; Vermeulen, P.B.; Preece, N.; Springer, C.J.; Powles, T.; et al. Preclinical evidence that trametinib enhances the response to antiangiogenic tyrosine kinase inhibitors in renal cell carcinoma. Mol. Cancer Ther. 2016, 15, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Meyer, C.; Zinner, R.; Meric-Bernstam, F.; Zahurak, M.L.; O’Connor, A.; Roszik, J.; Shaw, K.; Ludwig, J.A.; Kurzrock, R.; et al. Phase Ib/II Study of the safety and efficacy of combination therapy with multikinase VEGF inhibitor Pazopanib and MEK inhibitor Trametinib in advanced soft tissue sarcoma. Clin. Cancer Res. 2017, 23, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Naito, N.; Yoshida, A.; Morimoto, Y.; Ouchida, M.; Shimizu, K.; Beppu, Y. Establishment and characterization of a biphasic synovial sarcoma cell line, SYO-1. Cancer Lett. 2004, 204, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.B.; Barrott, J.J.; Xie, M.; Haldar, M.; Jin, H.; Zhu, J.F.; Monument, M.J.; Mosbruger, T.L.; Langer, E.M.; Randall, R.L.; et al. The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis. Oncogene 2016, 35, 5021–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renwick, P.J.; Reeves, B.R.; Dal Cin, P.; Fletcher, C.D.; Kempski, H.; Sciot, R.; Kazmierczak, B.; Jani, K.; Sonobe, H.; Knight, J.C. Two categories of synovial sarcoma defined by divergent chromosome translocation breakpoints in Xp11.2, with implications for the histologic sub-classification of synovial sarcoma. Cytogenet. Genome Res. 1995, 70, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, T.; Mezzelani, A.; Riva, C.; Fabbri, A.; Dal Bo, L.; Sampietro, G.; Perego, P.; Casali, P.; Zunino, F.; Sozzi, G.; et al. Analysis of SYT-SSX fusion transcripts and bcl-2 expression and phosphorylation status in synovial sarcoma. Lab. Investig. 2000, 80, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Mezzelani, A.; Mariani, L.; Tamborini, E.; Agus, V.; Riva, C.; Lo Vullo, S.; Fabbri, A.; Stumbo, M.; Azzarelli, A.; Casali, P.G.; et al. SYT-SSX fusion genes and prognosis in synovial sarcoma. Br. J. Cancer 2001, 85, 1535–1539. [Google Scholar] [CrossRef]
- Xie, Y.; Skytting, B.; Nilsson, G.; Gasbarri, A.; Haslam, K.; Bartolazzi, A.; Brodin, B.; Mandahl, N.; Larsson, O. SYT-SSX is Critical for cyclin D1 expression in synovial sarcoma cells: A gain of function of the t(X;18)(p11.2;q11.2) translocation. Cancer Res. 2002, 62, 3861–3867. [Google Scholar]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef]
- Kern, D.H.; Morgan, C.R.; Hildebrand-Zanki, S.U. In vitro pharmacodynamics of 1-beta-D-arabinofuranosylcytosine: Synergy of antitumor activity with cis-diamminedichloroplatinum(II). Cancer Res. 1988, 48, 117–121. [Google Scholar]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Cassinelli, G.; Lanzi, C.; Tortoreto, M.; Cominetti, D.; Petrangolini, G.; Favini, E.; Zaffaroni, N.; Pisano, C.; Penco, S.; Vlodavsky, I.; et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem. Pharmacol. 2013, 85, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Cuccuru, G.; Lanzi, C.; Cassinelli, G.; Pratesi, G.; Tortoreto, M.; Petrangolini, G.; Seregni, E.; Martinetti, A.; Laccabue, D.; Zanchi, C.; et al. Cellular effects and antitumor activity of RET inhibitor RPI-1 on MEN2A-associated medullary thyroid carcinoma. J. Natl. Cancer Inst. 2004, 96, 1006–1014. [Google Scholar] [CrossRef]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef]
- Miserocchi, G.; Mercatali, L.; Liverani, C.; De Vita, A.; Spadazzi, C.; Pieri, F.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ibrahim, T. Management and potentialities of primary cancer cultures in preclinical and translational studies. J. Transl. Med. 2017, 2017 15, 229. [Google Scholar] [CrossRef]
- Brodin, B.A.; Wennerberg, K.; Lidbrink, E.; Brosjö, O.; Potdar, S.; Wilson, J.N.; Ma, L.; Moens, L.N.; Hesla, A.; Porovic, E.; et al. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br. J. Cancer 2019, 120, 435–443. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanzi, C.; Dal Bo, L.; Favini, E.; Tortoreto, M.; Beretta, G.L.; Arrighetti, N.; Zaffaroni, N.; Cassinelli, G. Overactive IGF1/Insulin Receptors and NRASQ61R Mutation Drive Mechanisms of Resistance to Pazopanib and Define Rational Combination Strategies to Treat Synovial Sarcoma. Cancers 2019, 11, 408. https://doi.org/10.3390/cancers11030408
Lanzi C, Dal Bo L, Favini E, Tortoreto M, Beretta GL, Arrighetti N, Zaffaroni N, Cassinelli G. Overactive IGF1/Insulin Receptors and NRASQ61R Mutation Drive Mechanisms of Resistance to Pazopanib and Define Rational Combination Strategies to Treat Synovial Sarcoma. Cancers. 2019; 11(3):408. https://doi.org/10.3390/cancers11030408
Chicago/Turabian StyleLanzi, Cinzia, Laura Dal Bo, Enrica Favini, Monica Tortoreto, Giovanni Luca Beretta, Noemi Arrighetti, Nadia Zaffaroni, and Giuliana Cassinelli. 2019. "Overactive IGF1/Insulin Receptors and NRASQ61R Mutation Drive Mechanisms of Resistance to Pazopanib and Define Rational Combination Strategies to Treat Synovial Sarcoma" Cancers 11, no. 3: 408. https://doi.org/10.3390/cancers11030408