Phosphorylation-Dependent Intra-Domain Interaction of the Cx37 Carboxyl-Terminus Controls Cell Survival

Abstract
:1. Introduction
2. Results
2.1. Intra-Domain Interaction within the Cx37-CT Supports Cell Survival
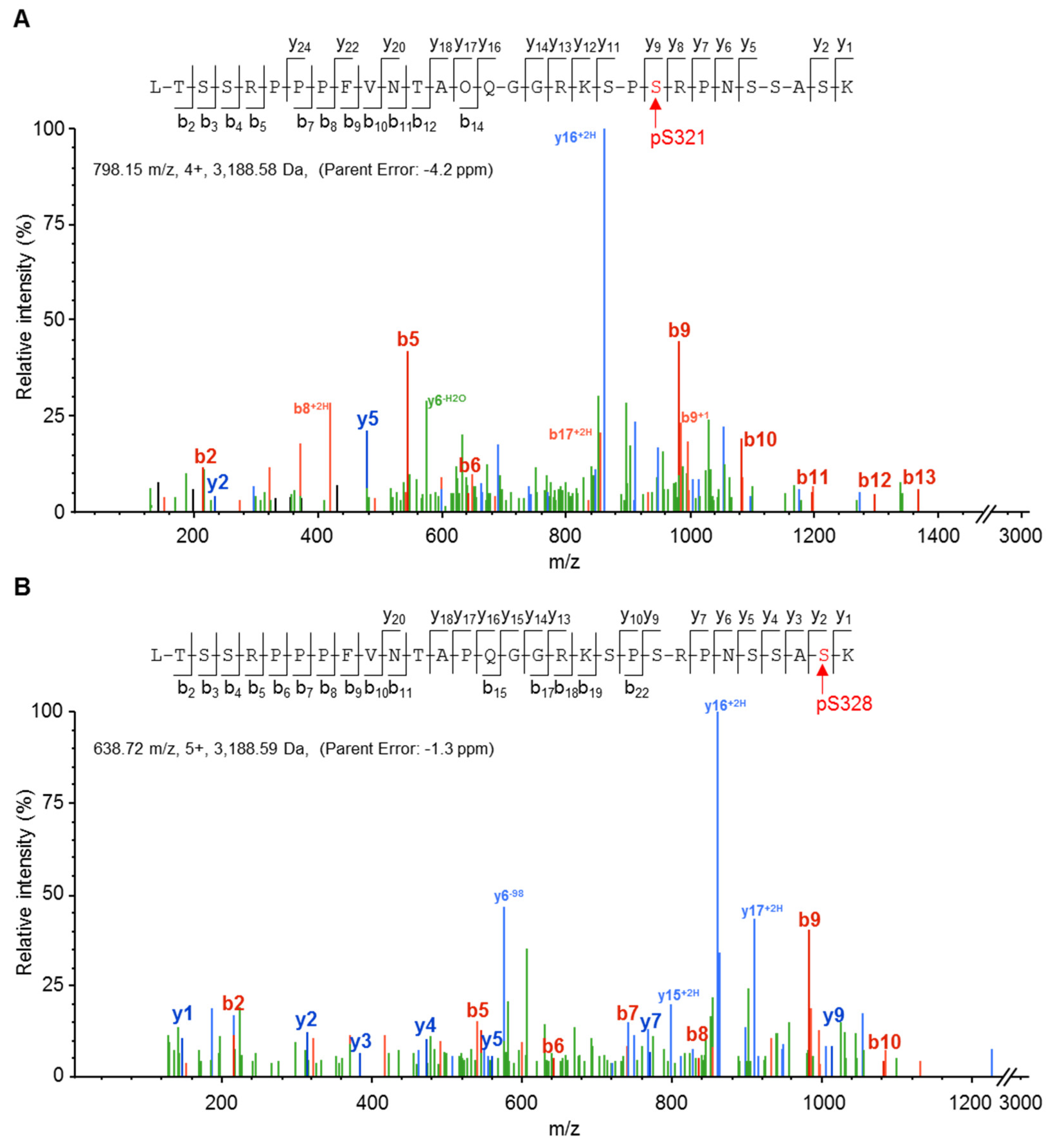
2.2. Cx37 Is a Multi-Phosphorylated Protein
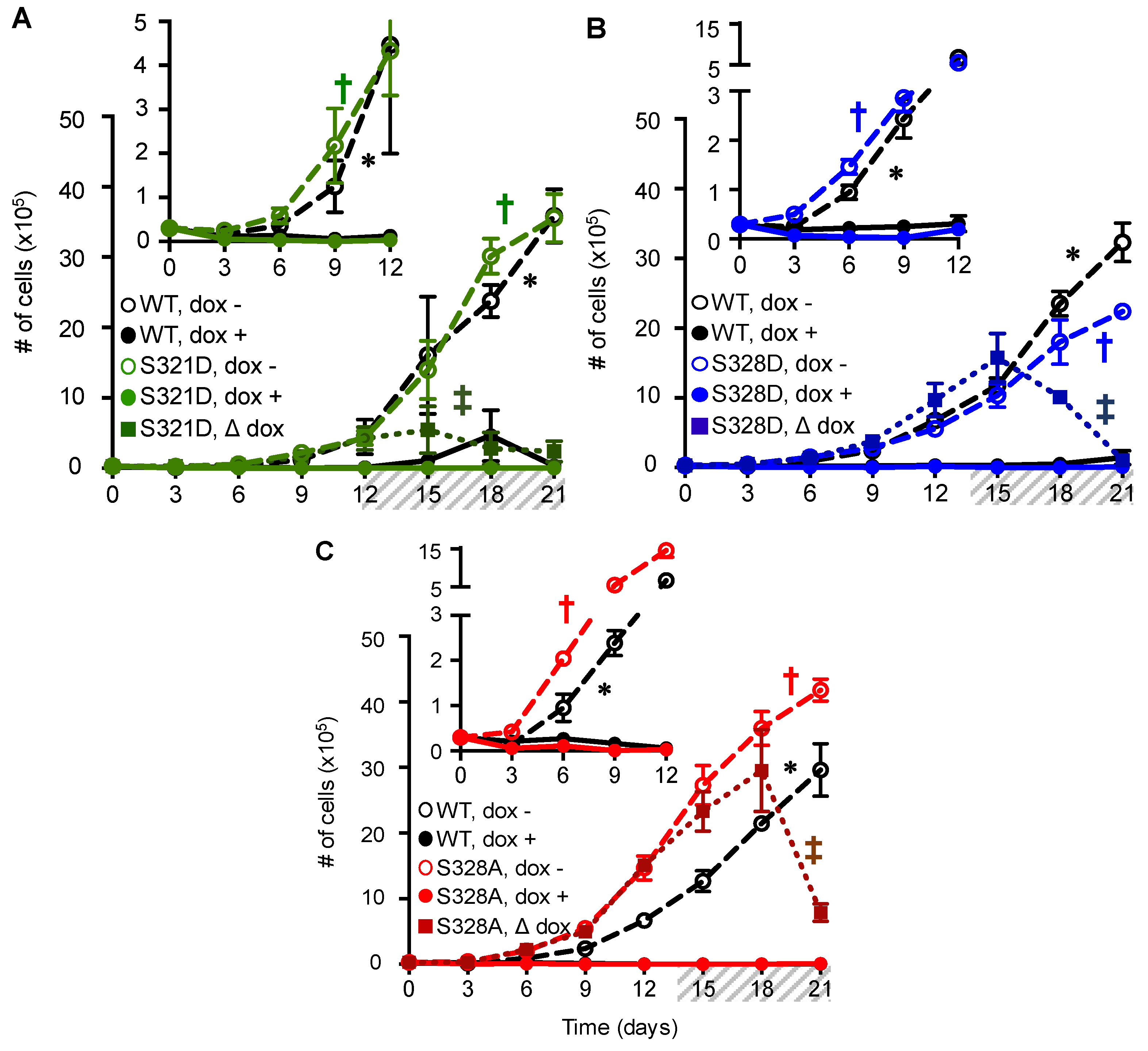
2.3. Phosphomimetic Mutations at Cx37-S321, S328, and S275 Induce Death of Rin Cells
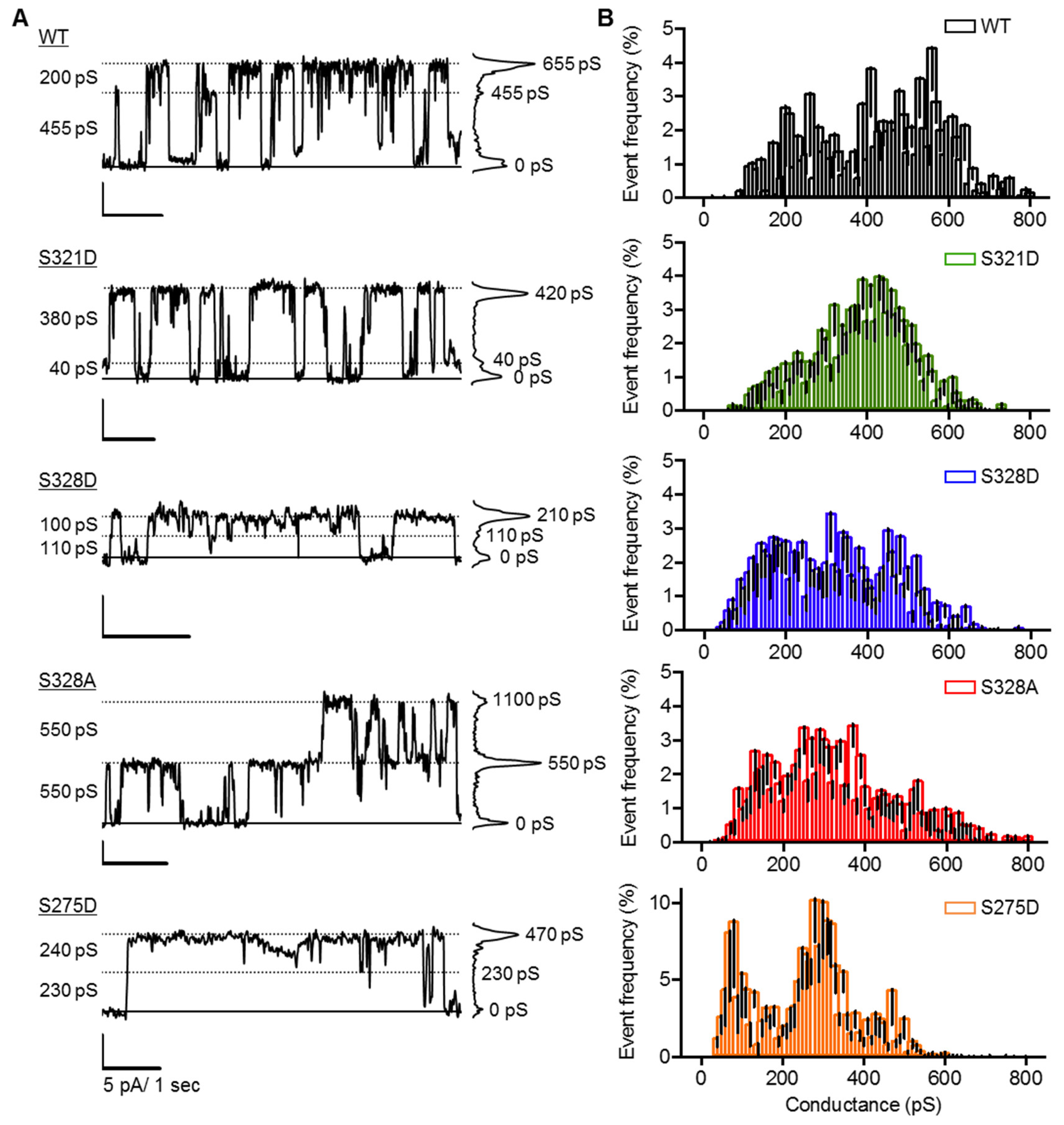
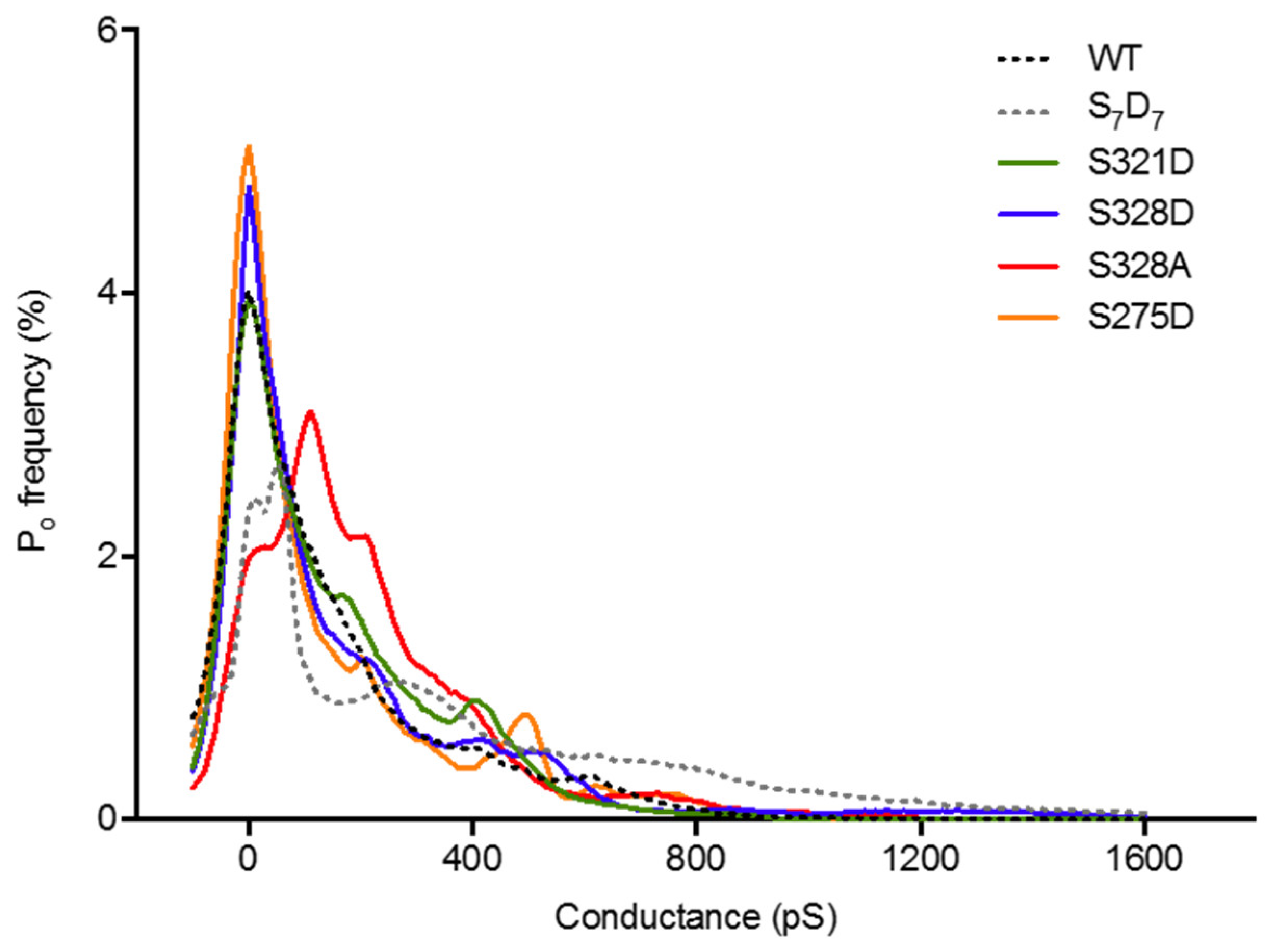
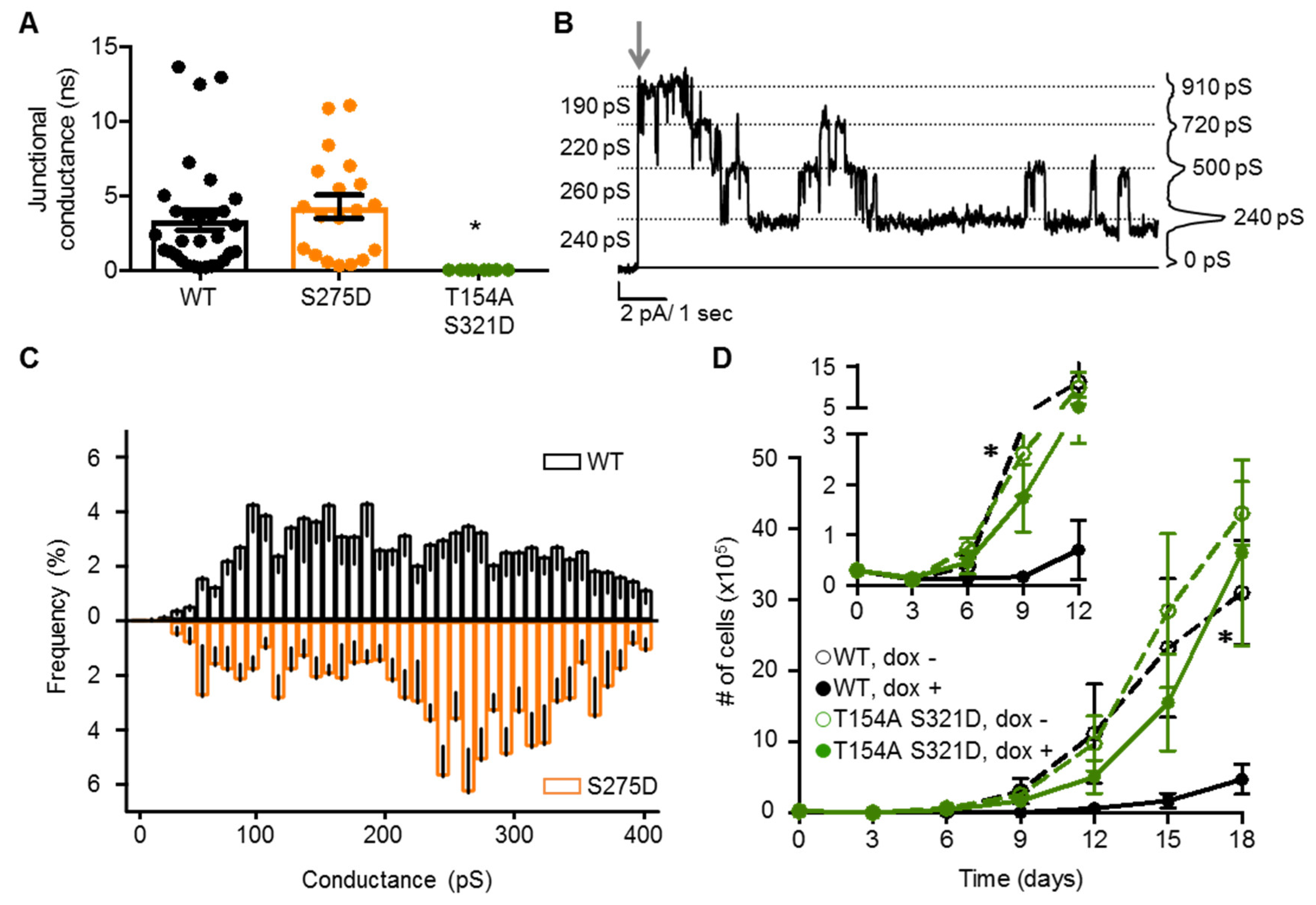
2.4. Neither Cx37 HCh Conductance nor Po Is Predictive of Cell Death
2.5. Channel Function Is Necessary for pCx37-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mutant Plasmid Generation, Transfection, and Cloning
4.3. Immunoblotting
4.4. Mass Spectrometry
4.5. Proliferation
4.6. Electrophysiology
4.6.1. GJCh Electrophysiology
4.6.2. HCh Electrophysiology
4.7. Fluorescence Activated Cell Sorting
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Igarashi, I.; Makino, T.; Suzuki, Y.; Kai, K.; Teranishi, M.; Takasaki, W.; Furuhama, K. Background lesions during a 24-month observation period in connexin 32-deficient mice. J. Vet. Med. Sci. 2013, 75, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Temme, A.; Buchmann, A.; Gabriel, H.D.; Nelles, E.; Schwarz, M.; Willecke, K. High incidence of spontaneous and chemically induced liver tumors in mice deficient for connexin32. Curr. Biol. 1997, 7, 713–716. [Google Scholar] [CrossRef]
- King, T.J.; Lampe, P.D. The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res. 2004, 64, 7191–7196. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.K.; Bechberger, J.F.; Welch, I.; Naus, C.C.; Laird, D.W. Cx26 knockout predisposes the mammary gland to primary mammary tumors in a DMBA-induced mouse model of breast cancer. Oncotarget 2015, 6, 37185–37199. [Google Scholar] [CrossRef] [PubMed]
- Cusato, K.; Ripps, H.; Zakevicius, J.; Spray, D.C. Gap junctions remain open during cytochrome c-induced cell death: Relationship of conductance to ‘bystander’ cell killing. Cell Death Differ. 2006, 13, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Nonaka, T.; Hamada, N.; Sakurai, H.; Hasegawa, M.; Funayama, T.; Kakizaki, T.; Kobayashi, Y.; Nakano, T. Heavy-ion-induced bystander killing of human lung cancer cells: Role of gap junctional intercellular communication. Cancer Sci. 2009, 100, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Krutovskikh, V.A.; Piccoli, C.; Yamasaki, H. Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene 2002, 21, 1989–1999. [Google Scholar] [CrossRef]
- Kameritsch, P.; Khandoga, N.; Pohl, U.; Pogoda, K. Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis. 2013, 4, e584. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef]
- Seul, K.H.; Kang, K.Y.; Lee, K.S.; Kim, S.H.; Beyer, E.C. Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem. Biophys. Res. Commun. 2004, 319, 1144–1151. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sanchez, H.A.; Veliz, L.P.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Rev. 2004, 47, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.P.; Rhee, S.K.; Lin, H.; Lal, R. Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J. Cell Biol. 2000, 148, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Contreras, J.E.; Sanchez, H.A.; Eugenin, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.; Saez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2002, 99, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Decrock, E.; Vanhaecke, T.; Leybaert, L.; Rogiers, V. Connexin43 signaling contributes to spontaneous apoptosis in cultures of primary hepatocytes. Toxicol. Sci. 2012, 125, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Lee, S.C.; Reuss, L.; Altenberg, G.A. Change in permeant size selectivity by phosphorylation of connexin 43 gap-junctional hemichannels by PKC. Proc. Natl. Acad. Sci. USA 2007, 104, 4919–4924. [Google Scholar] [CrossRef] [PubMed]
- Batra, N.; Riquelme, M.A.; Burra, S.; Kar, R.; Gu, S.; Jiang, J.X. Direct Regulation of Osteocytic Connexin 43 Hemichannels through AKT Kinase Activated by Mechanical Stimulation. J. Biol. Chem. 2014, 289, 10582–10591. [Google Scholar] [CrossRef]
- Li, H.; Liu, T.F.; Lazrak, A.; Peracchia, C.; Goldberg, G.S.; Lampe, P.D.; Johnson, R.G. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 1996, 134, 1019–1030. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kam, Y.; Koo, S.K.; Joe, C.O. Gating connexin 43 channels reconstituted in lipid vesicles by mitogen-activated protein kinase phosphorylation. J. Biol. Chem. 1999, 274, 5581–5587. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.L.; Pontifex, T.K.; Li, H.; Solan, J.L.; Lampe, P.D.; Sorgen, P.L.; Burt, J.M. Regulation of Cx37 channel and growth-suppressive properties by phosphorylation. J. Cell Sci. 2017, 130, 3308–3321. [Google Scholar] [CrossRef] [PubMed]
- Good, M.E.; Nelson, T.K.; Simon, A.M.; Burt, J.M. A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 2011, 124, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.K.; Sorgen, P.L.; Burt, J.M. Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am. J. Physiol. Cell Physiol. 2013, 305, C1246–C1256. [Google Scholar] [CrossRef] [PubMed]
- Burt, J.M.; Nelson, T.K.; Simon, A.M.; Fang, J.S. Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1103–C1112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, Y.; Lin, X.; Wong, H.C.; Xu, Q.; Jiang, J.; Wang, S.; Lurtz, M.M.; Louis, C.F.; Veenstra, R.D.; et al. Molecular interaction and functional regulation of connexin50 gap junctions by calmodulin. Biochem. J. 2011, 435, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.M.; Seul, K.H.; Berthoud, V.M.; Lau, A.F.; Sagar, G.D.; Beyer, E.C. Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell Biol. Res. Commun. 2000, 3, 115–121. [Google Scholar] [CrossRef]
- Traub, O.; Hertlein, B.; Kasper, M.; Eckert, R.; Krisciukaitis, A.; Hulser, D.; Willecke, K. Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human HeLa cells. Eur. J. Cell Biol. 1998, 77, 313–322. [Google Scholar] [CrossRef]
- Kopanic, J.L.; Al-mugotir, M.H.; Kieken, F.; Zach, S.; Trease, A.J.; Sorgen, P.L. Characterization of the connexin45 carboxyl-terminal domain structure and interactions with molecular partners. Biophys. J. 2014, 106, 2184–2195. [Google Scholar] [CrossRef]
- Good, M.E.; Ek-Vitorin, J.F.; Burt, J.M. Extracellular loop cysteine mutant of cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 2012, 245, 369–380. [Google Scholar] [CrossRef]
- Belousov, A.B.; Fontes, J.D.; Freitas-Andrade, M.; Naus, C.C. Gap junctions and hemichannels: Communicating cell death in neurodevelopment and disease. BMC Cell Biol. 2017, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Worsdorfer, P.; Wagner, N.; Ergun, S. The role of connexins during early embryonic development: Pluripotent stem cells, gene editing, and artificial embryonic tissues as tools to close the knowledge gap. Histochem. Cell Biol. 2018, 150, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.R.; Ferdinandy, P.; Beyer, E.C.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Czyz, J.; Piwowarczyk, K.; Paw, M.; Luty, M.; Wrobel, T.; Catapano, J.; Madeja, Z.; Ryszawy, D. Connexin-dependent intercellular stress signaling in tissue homeostasis and tumor development. Acta Biochim. Pol. 2017, 64, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D.; Green, C.R. Connexin43 in retinal injury and disease. Prog. Retin. Eye Res. 2016, 51, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Kruger, O.; Plum, A.; Kim, J.S.; Winterhager, E.; Maxeiner, S.; Hallas, G.; Kirchhoff, S.; Traub, O.; Lamers, W.H.; Willecke, K. Defective vascular development in connexin 45-deficient mice. Development 2000, 127, 4179–4193. [Google Scholar] [PubMed]
- Fang, J.S.; Coon, B.G.; Gillis, N.; Chen, Z.; Qiu, J.; Chittenden, T.W.; Burt, J.M.; Schwartz, M.A.; Hirschi, K.K. Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun. 2017, 8, 2149. [Google Scholar] [CrossRef]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial cell apoptosis in angiogenesis and vessel regression. Cell. Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef]
- Fang, J.S.; Angelov, S.N.; Simon, A.M.; Burt, J.M. Cx40 is required for, and cx37 limits, postischemic hindlimb perfusion, survival and recovery. J. Vasc. Res. 2012, 49, 2–12. [Google Scholar] [CrossRef]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef]
- Li, H.; Wang, F. The role of connexin43 in diabetic microvascular complications. Discov. Med. 2016, 22, 275–280. [Google Scholar] [PubMed]
- Mugisho, O.O.; Green, C.R.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannels: A potential drug target for the treatment of diabetic retinopathy. Drug Discov. Today 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Batra, N.; Kar, R.; Jiang, J.X. Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim. Biophys. Acta 2012, 1818, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.C.; Gouw, J.W.; Naus, C.C.; Foster, L.J. Connexin multi-site phosphorylation: Mass spectrometry-based proteomics fills the gap. Biochim. Biophys. Acta 2013, 1828, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Spagnol, G.; Pontifex, T.K.; Burt, J.M.; Sorgen, P.L. Chemical shift assignments of the connexin37 carboxyl terminal domain. Biomol. NMR Assign. 2017, 11, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef]
- Sorgen, P.L.; Duffy, H.S.; Sahoo, P.; Coombs, W.; Delmar, M.; Spray, D.C. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J. Biol. Chem. 2004, 279, 54695–54701. [Google Scholar] [CrossRef]
- Doble, B.W.; Dang, X.; Ping, P.; Fandrich, R.R.; Nickel, B.E.; Jin, Y.; Cattini, P.A.; Kardami, E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004, 117, 507–514. [Google Scholar] [CrossRef]
- Simon, A.M.; Chen, H.; Jackson, C.L. Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun. Adhes. 2006, 13, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Krantz, J.; Barker, N.; Coletta, R.L.; Rafikov, R.; Luo, M.; Hojlund, K.; Mandarino, L.J.; Langlais, P.R. Characterization of the CLASP2 Protein Interaction Network Identifies SOGA1 as a Microtubule-Associated Protein. Mol. Cell. Proteom. 2017, 16, 1718–1735. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Cote, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, D1063–D1069. [Google Scholar] [CrossRef] [PubMed]
- Kurjiaka, D.T.; Steele, T.D.; Olsen, M.V.; Burt, J.M. Gap junction permeability is diminished in proliferating vascular smooth muscle cells. Am. J. Physiol. 1998, 275, C1674–C1682. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Detected | Phosphorylated 1 |
|---|---|---|
| S275 | 33 | 5 |
| S285 | 57 | 0 |
| S302 | 559 | 0 |
| S319 | 559 | 204 |
| S321 | 559 | 4 |
| S325 | 559 | 2 |
| S328 | 559 | 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacobsen, N.L.; Pontifex, T.K.; Langlais, P.R.; Burt, J.M. Phosphorylation-Dependent Intra-Domain Interaction of the Cx37 Carboxyl-Terminus Controls Cell Survival. Cancers 2019, 11, 188. https://doi.org/10.3390/cancers11020188
Jacobsen NL, Pontifex TK, Langlais PR, Burt JM. Phosphorylation-Dependent Intra-Domain Interaction of the Cx37 Carboxyl-Terminus Controls Cell Survival. Cancers. 2019; 11(2):188. https://doi.org/10.3390/cancers11020188
Chicago/Turabian StyleJacobsen, Nicole L., Tasha K. Pontifex, Paul R. Langlais, and Janis M. Burt. 2019. "Phosphorylation-Dependent Intra-Domain Interaction of the Cx37 Carboxyl-Terminus Controls Cell Survival" Cancers 11, no. 2: 188. https://doi.org/10.3390/cancers11020188