The Benzimidazole-Based Anthelmintic Parbendazole: A Repurposed Drug Candidate That Synergizes with Gemcitabine in Pancreatic Cancer

 , , ,
, , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
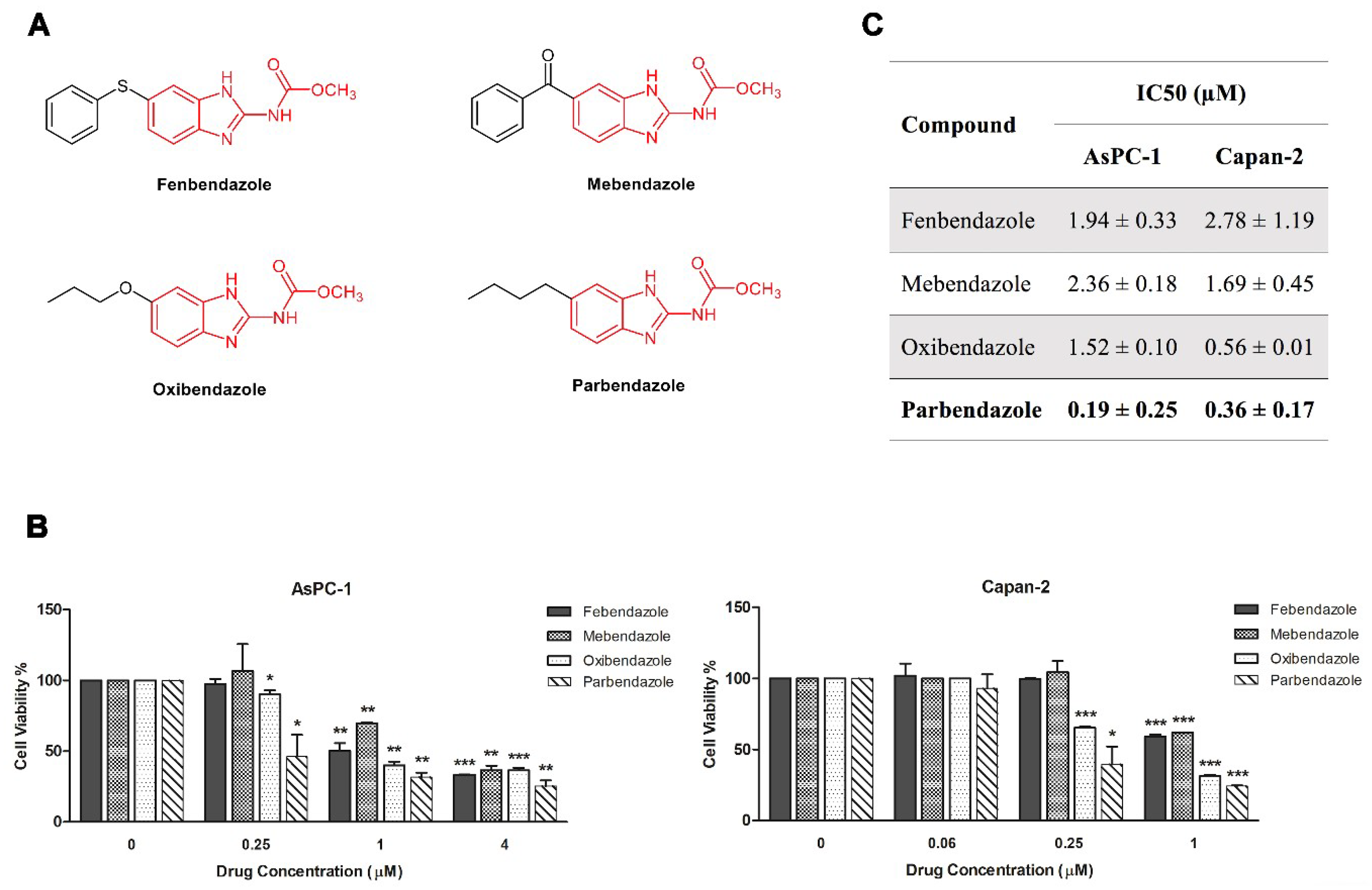
2.1. Parbendazole Has the Lowest IC50 in the Panel of Tested Benzimidazoles
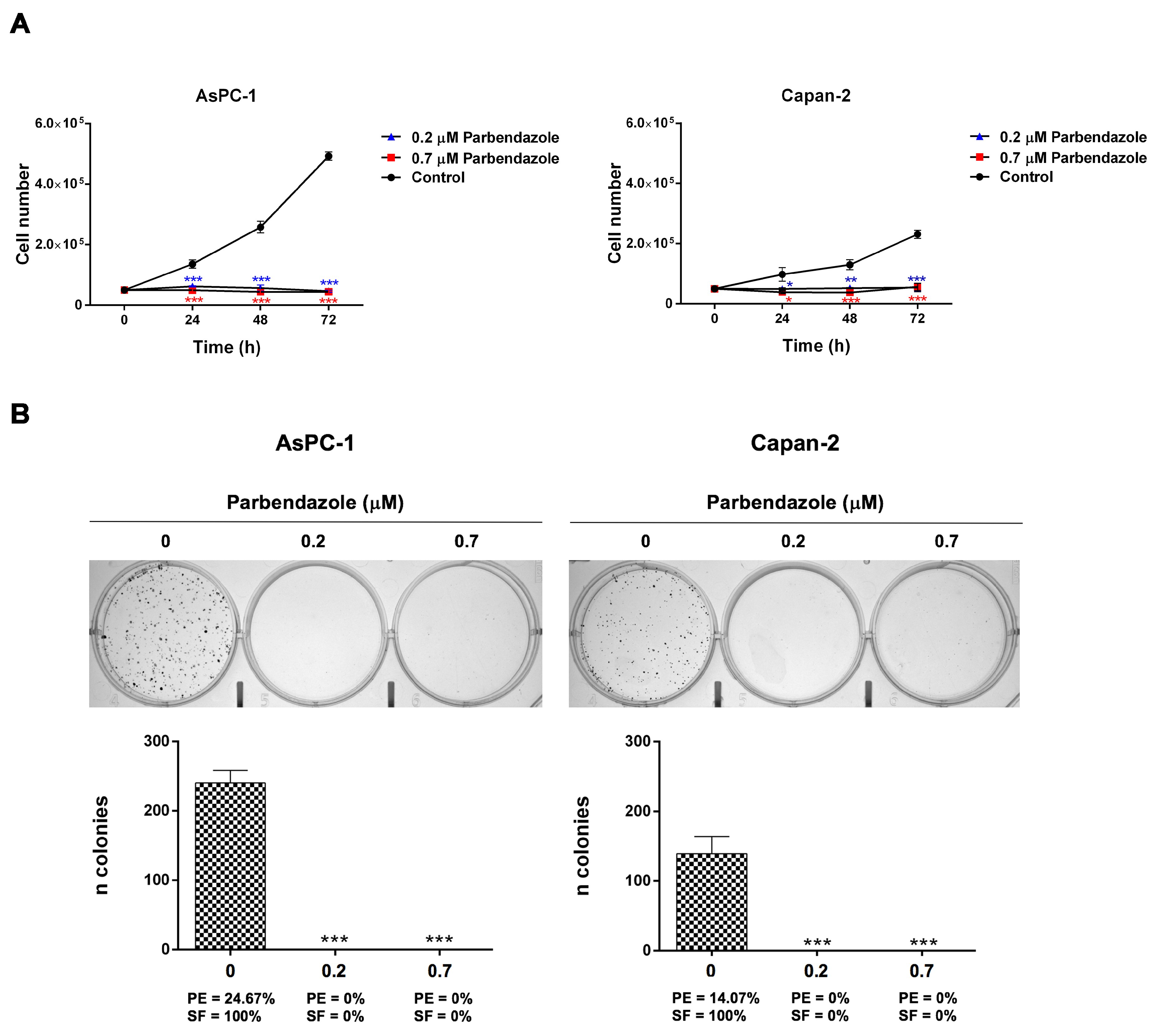
2.2. Parbendazole Hampers Growth and Clonogenicity of PC Cell Lines
2.3. Parbendazole Alters Mitotic Spindles Formation in PC Cells
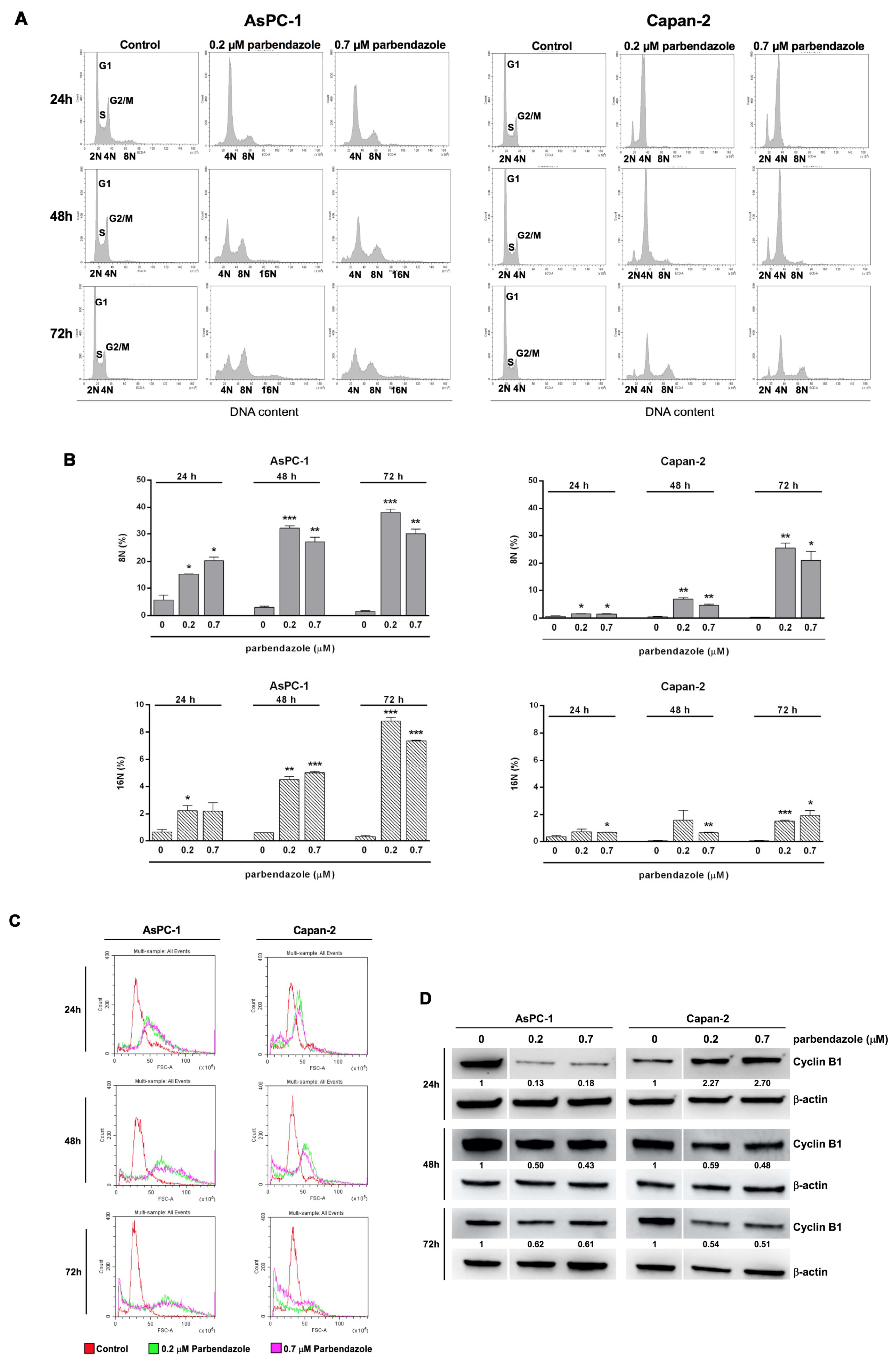
2.4. Parbendazole Affects Cell Cycle Altering DNA Content and Size of PC Cells
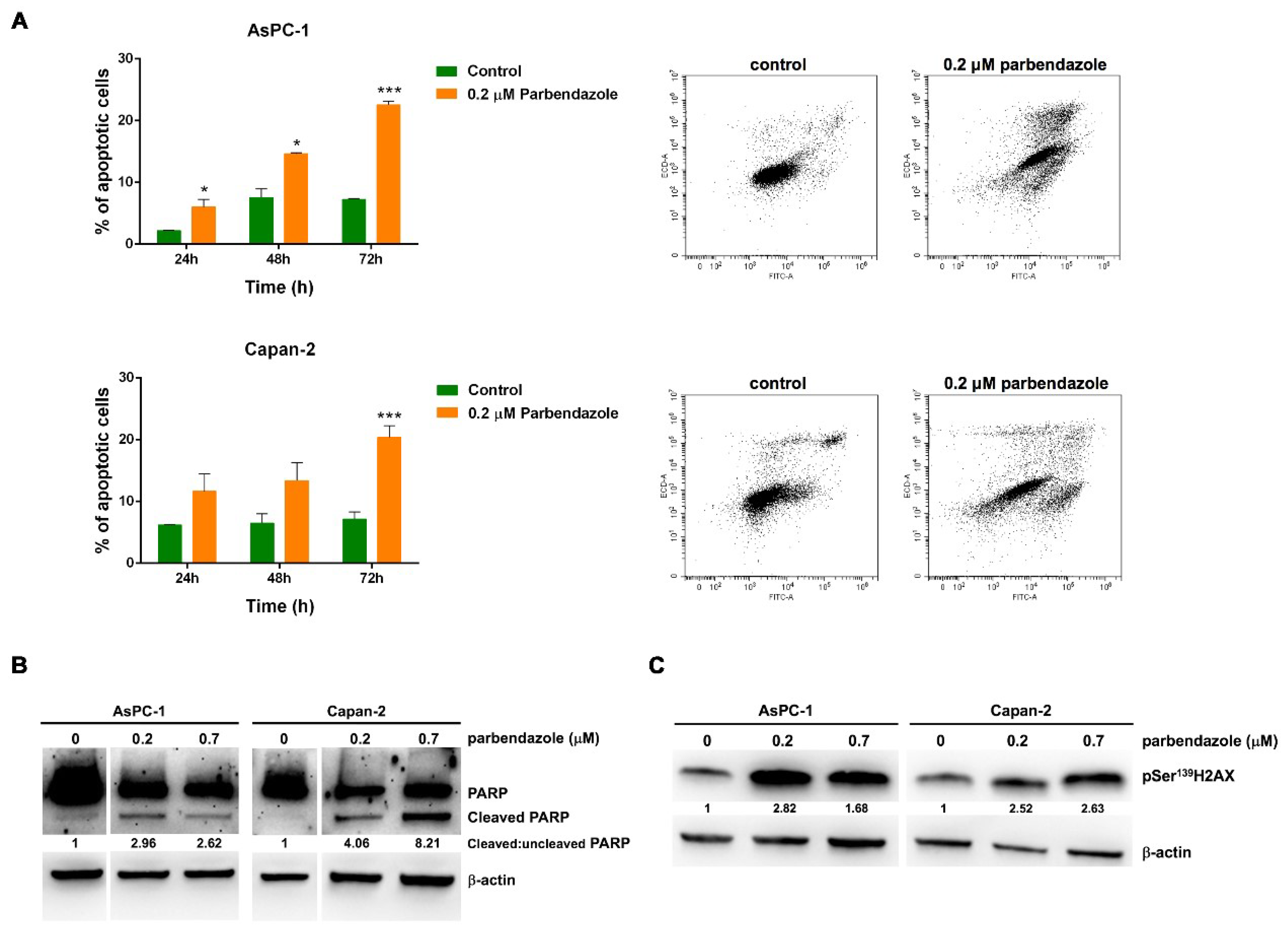
2.5. Parbendazole Promotes Apoptosis and DNA Damage in PC Cells
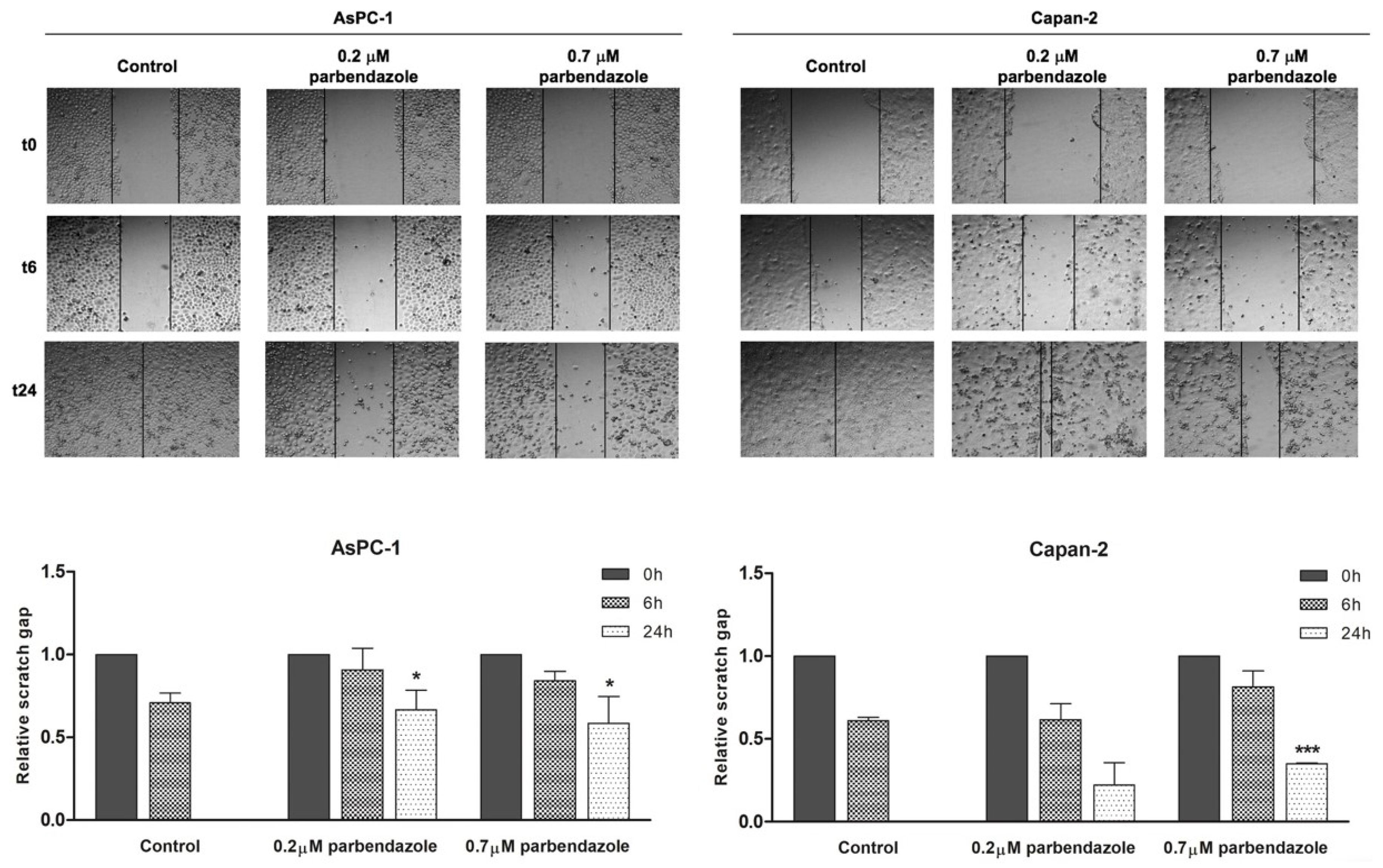
2.6. Parbendazole Impairs PC Cell Migration
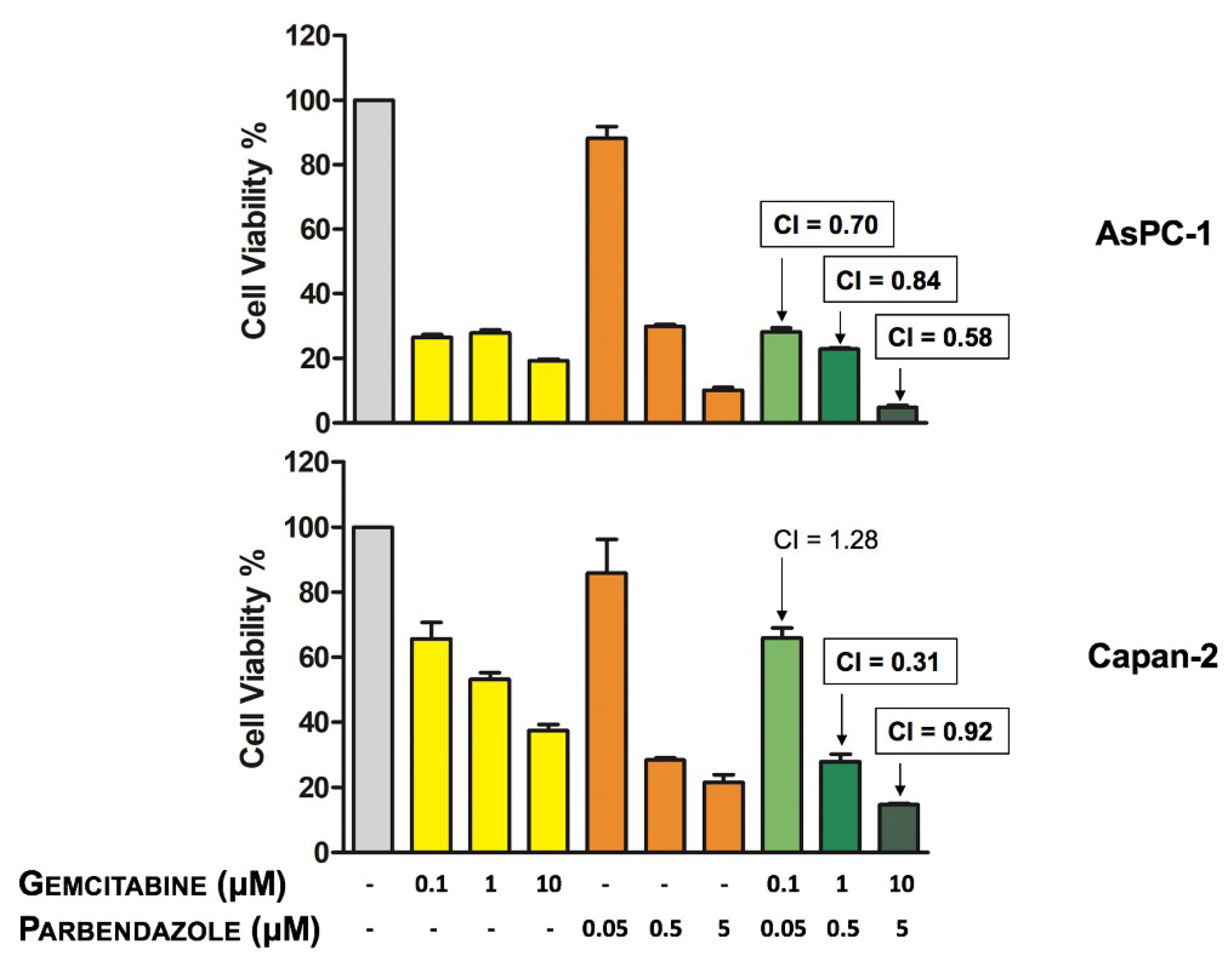
2.7. Combinations of Parbendazole and Gemcitabine Synergistically Affect PC Cell Viability
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Lines and Chemicals
4.3. Cell Viability and Cell Growth Assays
4.4. Colony Formation Assay
4.5. Immunofluorescence
4.6. Cell Cycle Analysis
4.7. Apoptosis Assay
4.8. Western Blotting Analysis
4.9. Wound-Healing Assay
4.10. Calculation of IC50 and Combination Index (CI)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renz, B.W.; D’Haese, J.G.; Werner, J.; Westphalen, C.B.; Ilmer, M. Repurposing established compounds to target pancreatic cancer stem cells (CSCs). Med. Sci. (Basel) 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilmer, M.; Westphalen, C.B.; Niess, H.; D’Haese, J.G.; Angele, M.K.; Werner, J.; Renz, B.W. Repurposed drugs in pancreatic ductal adenocarcinoma: An update. Cancer J. 2019, 25, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total neoadjuvant therapy with FOLFIRINOX in combination with losartan followed by chemoradiotherapy for locally advanced pancreatic cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Veschi, S.; De Lellis, L.; Florio, R.; Lanuti, P.; Massucci, A.; Tinari, N.; De Tursi, M.; di Sebastiano, P.; Marchisio, M.; Natoli, C.; et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, e236. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Laghezza, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Tortorella, P.; Veschi, S.; et al. Synthesis of novel benzothiazole amides: Evaluation of PPAR activity and anti-proliferative effects in paraganglioma, pancreatic and colorectal cancer cell lines. Bioorg. Med. Chem. Lett. 2019, 29, 2302–2306. [Google Scholar] [CrossRef]
- De Filippis, B.; De Lellis, L.; Florio, R.; Ammazzalorso, A.; Amoia, P.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R.; Veschi, S.; et al. Synthesis and cytotoxic effects on pancreatic cancer cells of resveratrol analogs. Med. Chem. Res. 2019, 28, 984–991. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P.; Vikas, P. The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience 2014, 8, e442. [Google Scholar] [CrossRef]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Hanusova, V.; Skalova, L.; Kralova, V.; Matouskova, P. Potential anti-cancer drugs commonly used for other indications. Curr. Cancer Drug Targets 2015, 15, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Feldmeier, H.; Bienzle, U.; Döhring, E.; Dietrich, M. Flubendazole versus mebendazole in intestinal helminthic infections. Acta Trop. 1982, 39, 185–189. [Google Scholar]
- Muser, R.K.; Paul, J.W. Safety of fenbendazole use in cattle. Mod. Vet. Pract. 1984, 65, 371–374. [Google Scholar]
- Horton, R.J. Albendazole in treatment of human cystic echinococcosis: 12 years of experience. Acta Trop. 1997, 64, 79–93. [Google Scholar] [CrossRef]
- Čáňová, K.; Rozkydalová, L.; Vokurková, D.; Rudolf, E. Flubendazole induces mitotic catastrophe and apoptosis in melanoma cells. Toxicol. In Vitro 2018, 46, 313–322. [Google Scholar] [CrossRef]
- He, L.; Shi, L.; Du, Z.; Huang, H.; Gong, R.; Ma, L.; Chen, L.; Gao, S.; Lyu, J.; Gu, H. Mebendazole exhibits potent anti-leukemia activity on acute myeloid leukemia. Exp. Cell Res. 2018, 369, 61–68. [Google Scholar] [CrossRef]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.J.; Zheng, F.M.; Xu, J.; Xu, L.Z.; Long, Z.J.; et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.R.; Bai, R.Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.S.; Kviecinski, M.R.; Ourique, F.; Parisotto, E.B.; Grinevicius, V.M.; Correia, J.F.; Wilhelm Filho, D.; Pedrosa, R.C. Albendazole as a promising molecule for tumor control. Redox Biol. 2016, 10, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Králová, V.; Hanušová, V.; Rudolf, E.; Čáňová, K.; Skálová, L. Flubendazole induces mitotic catastrophe and senescence in colon cancer cells in vitro. J. Pharm. Pharmacol. 2016, 68, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Varbanov, H.P.; Kuttler, F.; Banfi, D.; Turcatti, G.; Dyson, P.J. Repositioning approved drugs for the treatment of problematic cancers using a screening approach. PLoS ONE 2017, 12, e0171052. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, J.; Pradhan, R.; Dandawate, P.R.; Subramaniam, D.; Ponnurangam, S.; Martis, E.A.F.; Ranpise, N.; Coutinho, E.C.; Khan, E.M.; Padhye, S.; et al. Spectral and molecular modeling studies on the influence of β-cyclodextrin and its derivatives on albendazole and its anti-proliferative activity Against pancreatic cancer cells. J. Pharm. Sci. Pharmacol. 2017, 3, 1–14. [Google Scholar] [CrossRef]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers (Basel) 2019, 11, 1284. [Google Scholar] [CrossRef] [Green Version]
- Dogra, N.; Kumar, A.; Mukhopadhyay, T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, e11926. [Google Scholar] [CrossRef]
- Iyer, P.; Makris, S. Developmental and Reproductive Toxicology of Pesticides. In Hayes’ Handbook of Pesticide Toxicology, 3rd ed.; Krieger, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 1, pp. 381–440. [Google Scholar]
- Al-Hadiya, B.M.H. Parbendazole. In Profiles of Drug Substances, Excipients and Related Methodology, 1st ed.; Brittain, H.G., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 35, pp. 263–284. [Google Scholar]
- Huang, Y.T.; Huang, D.M.; Guh, J.H.; Chen, I.L.; Tzeng, C.C.; Teng, C.M. CIL-102 interacts with microtubule polymerization and causes mitotic arrest following apoptosis in the human prostate cancer PC-3 cell line. J. Biol. Chem. 2005, 280, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.C.; Senese, S.; France, B.; Gholkar, A.A.; Damoiseaux, R.; Torres, J.Z. Computational cell cycle profiling of cancer cells for prioritizing FDA-approved drugs with repurposing potential. Sci. Rep. 2017, 7, e11261. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. Microtubules in cell migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Ansari, D.; Tingstedt, B.; Andersson, B.; Holmquist, F.; Sturesson, C.; Williamsson, C.; Sasor, A.; Borg, D.; Bauden, M.; Andersson, R. Pancreatic cancer: Yesterday, today and tomorrow. Future Oncol. 2016, 12, 1929–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awaji, M.; Singh, R.K. Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers (Basel) 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β signaling activation by mast cells contribute to resistance to gemcitabine/nabpaclitaxel in pancreatic cancer. Cancers (Basel) 2019, 11, 330. [Google Scholar] [CrossRef] [Green Version]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [Green Version]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Kienitz, A.; Hofmann, I.; Müller, R.; Bastians, H. Crosstalk of the mitotic spindle assembly checkpoint with p53 to prevent polyploidy. Oncogene 2004, 23, 6845–6853. [Google Scholar] [CrossRef] [Green Version]
- Sampson, V.B.; Vetter, N.S.; Zhang, W.; Patil, P.U.; Mason, R.W.; George, E.; Gorlick, R.; Kolb, E.A. Integrating mechanisms of response and resistance against the tubulin binding agent Eribulin in preclinical models of osteosarcoma. Oncotarget 2016, 7, 86594–86607. [Google Scholar] [CrossRef] [Green Version]
- Wawro, M.E.; Chojnacka, K.; Wieczorek-Szukała, K.; Sobierajska, K. Niewiarowska, Invasive colon cancer cells induce transdifferentiation of endothelium to cancer-associated fibroblasts through microtubules enriched in tubulin-β3. Int. J. Mol. Sci. 2019, 20, 53. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, G.; Henriksson, J.; Sander, B.; Wright, A.P. Mixed-species RNAseq analysis of human lymphoma cells adhering to mouse stromal cells identifies a core gene set that is also differentially expressed in the lymph node microenvironment of mantle cell lymphoma and chronic lymphocytic leukemia patients. Haematologica 2018, 103, 666–678. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P., IV; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Driscoll, D.R.; DeJesus-Monge, W.E.; Quattrochi, B.; Appleman, V.A.; Ou, J.; Zhu, L.J.; Yoshida, N.; Yamazaki, S.; Takayama, T.; et al. Activation of WNT/β-catenin signaling enhances pancreatic cancer development and the malignant potential via up-regulation of Cyr61. Neoplasia 2016, 18, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene expression comparison between the lymph node-positive and -negative reveals a peculiar immune microenvironment signature and a theranostic role for WNT targeting in pancreatic ductal adenocarcinoma: A pilot study. Cancers (Basel) 2019, 11, 942. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, G.G.; Cook, T.A. Microtubules and signal transduction. Curr. Opin. Cell Biol. 1999, 11, 81–94. [Google Scholar] [CrossRef]
- Arend, R.C.; Londoño-Joshi, A.I.; Gangrade, A.; Katre, A.A.; Kurpad, C.; Li, Y.; Samant, R.S.; Li, P.K.; Landen, C.N.; Yang, E.S.; et al. Niclosamide and its analogs are potent inhibitors of Wnt/β-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget 2016, 7, 86803–86815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.L.; Lian, D.D.; Zhu, M.J.; Li, X.M.; Lee, J.K.; Yoon, T.J.; Lee, J.H.; Jiang, R.H.; Kim, C.D. Antitumor effect of albendazole on cutaneous squamous cell carcinoma (SCC) cells. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Yashiro, M. Molecular targets for the treatment of pancreatic cancer: Clinical and experimental studies. World J. Gastroenterol. 2016, 22, 776–789. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Matters, G.L.; McGovern, C.; Harms, J.F.; Markovic, K.; Anson, K.; Jayakumar, C.; Martenis, M.; Awad, C.; Smith, J.P. Role of endogenous cholecystokinin on growth of human pancreatic cancer. Int. J. Oncol. 2011, 38, 593–601. [Google Scholar]
- Smith, J.P.; Solomon, T.E. Cholecystokinin and pancreatic cancer: The chicken or the egg? Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, e6744. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ma, Y.; Shankar, S.; Srivastava, R.K. Role of SATB2 in human pancreatic cancer: Implications in transformation and a promising biomarker. Oncotarget 2016, 7, 57783–57797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florio, R.; De Lellis, L.; Veschi, S.; Verginelli, F.; di Giacomo, V.; Gallorini, M.; Perconti, S.; Sanna, M.; Mariani-Costantini, R.; Natale, A.; et al. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci. Rep. 2018, 8, e13610. [Google Scholar] [CrossRef] [PubMed]
- De Winter, J.C.F. Using the student’s “t”-test with extremely small sample sizes. Pract. Assess. Res. Eval. 2013, 18. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florio, R.; Veschi, S.; di Giacomo, V.; Pagotto, S.; Carradori, S.; Verginelli, F.; Cirilli, R.; Casulli, A.; Grassadonia, A.; Tinari, N.; et al. The Benzimidazole-Based Anthelmintic Parbendazole: A Repurposed Drug Candidate That Synergizes with Gemcitabine in Pancreatic Cancer. Cancers 2019, 11, 2042. https://doi.org/10.3390/cancers11122042
Florio R, Veschi S, di Giacomo V, Pagotto S, Carradori S, Verginelli F, Cirilli R, Casulli A, Grassadonia A, Tinari N, et al. The Benzimidazole-Based Anthelmintic Parbendazole: A Repurposed Drug Candidate That Synergizes with Gemcitabine in Pancreatic Cancer. Cancers. 2019; 11(12):2042. https://doi.org/10.3390/cancers11122042
Chicago/Turabian StyleFlorio, Rosalba, Serena Veschi, Viviana di Giacomo, Sara Pagotto, Simone Carradori, Fabio Verginelli, Roberto Cirilli, Adriano Casulli, Antonino Grassadonia, Nicola Tinari, and et al. 2019. "The Benzimidazole-Based Anthelmintic Parbendazole: A Repurposed Drug Candidate That Synergizes with Gemcitabine in Pancreatic Cancer" Cancers 11, no. 12: 2042. https://doi.org/10.3390/cancers11122042