Aberrant DNA Methylation Predicts Melanoma-Specific Survival in Patients with Acral Melanoma

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Clinicopathologic Features
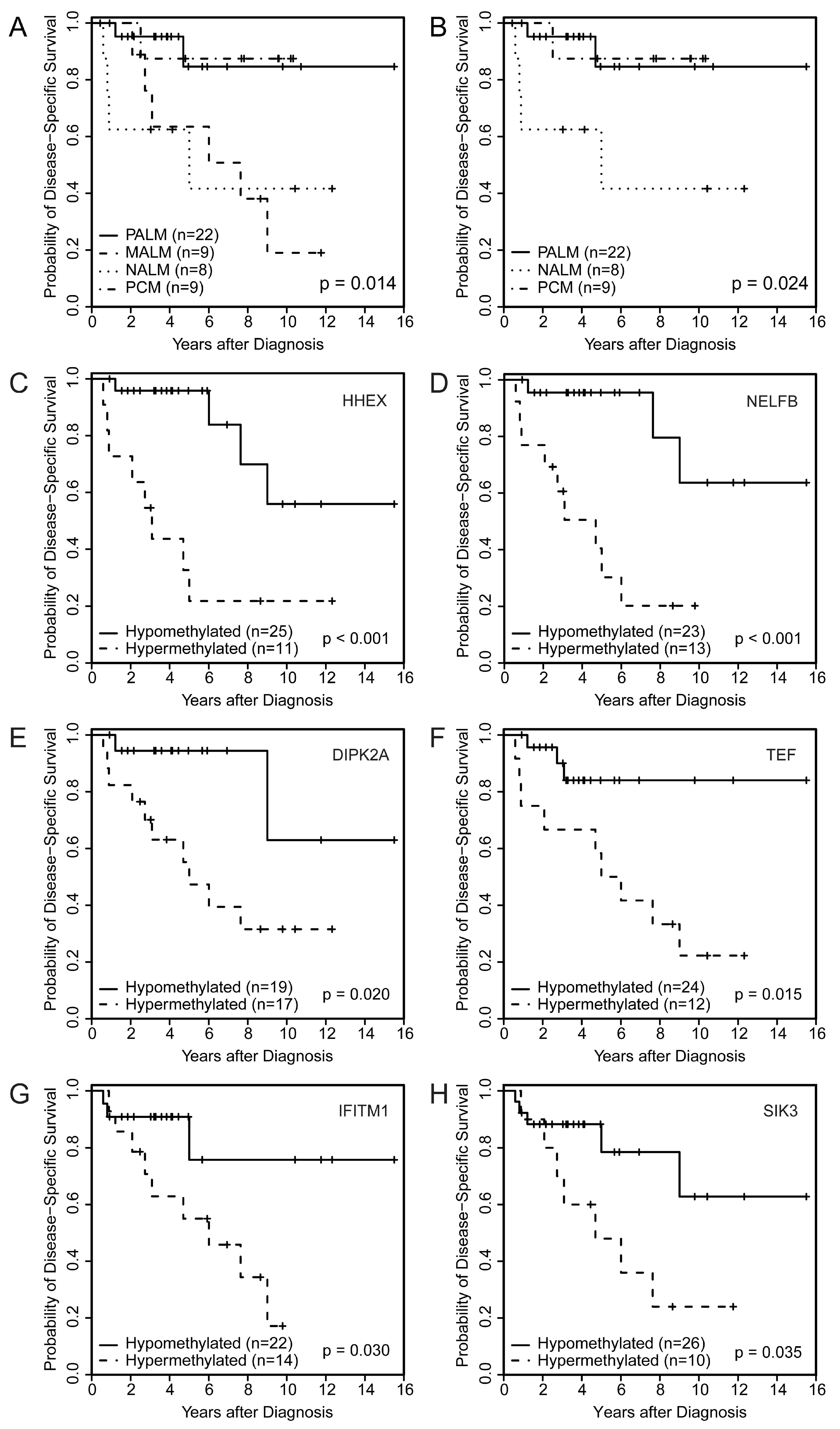
2.2. Association of Melanoma Subtype with Clinicopathologic Parameters, Genetic Alterations, and Outcome
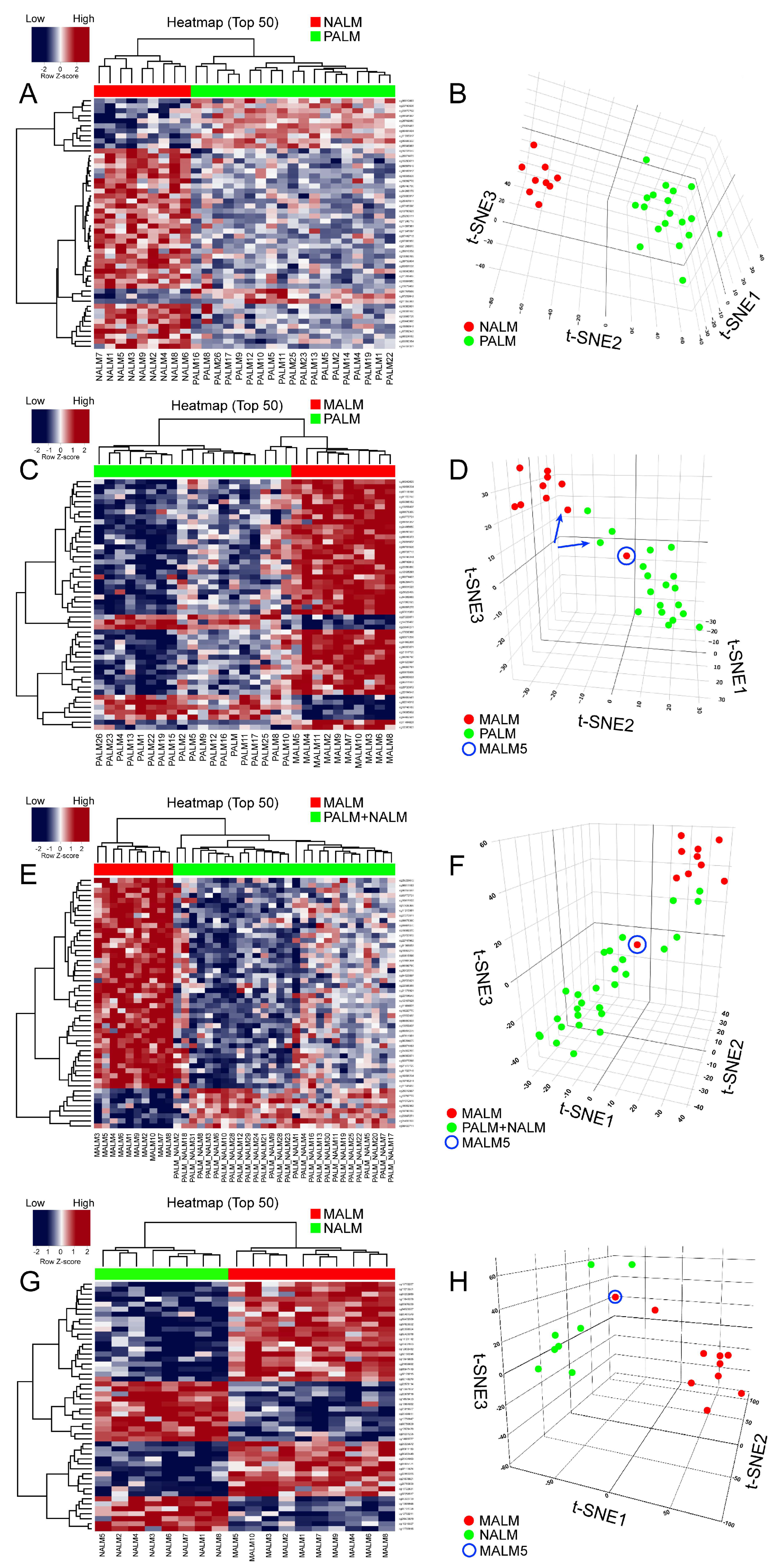
2.3. Performance of the Methylome Signature in Distinguishing Malignant from Benign Melanocytic Neoplasms
2.4. Identification of a Primary Acral Lentiginous Melanoma (PALM)-Specific Methylome Signature
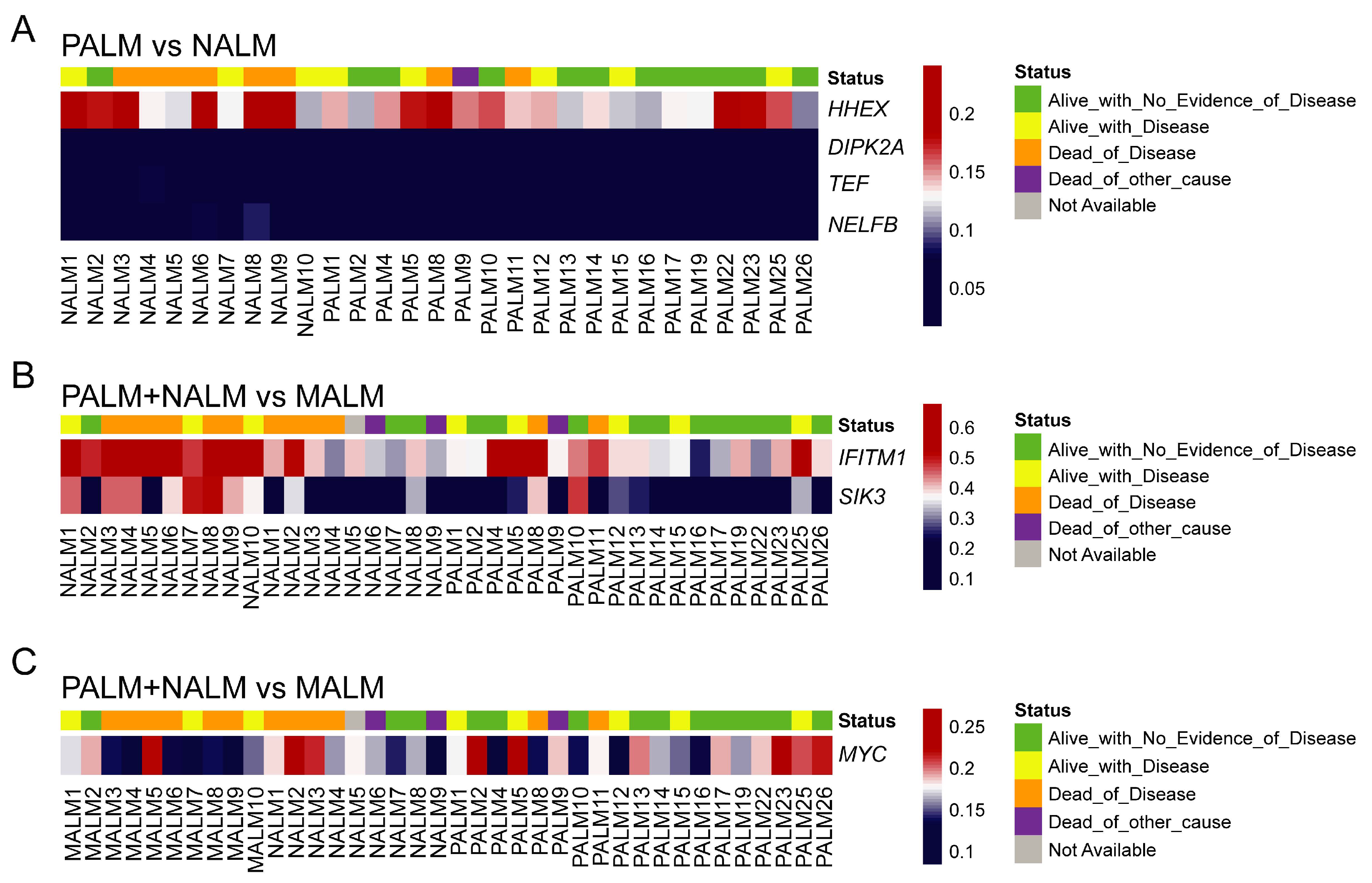
2.5. Association of PALM-Specific Methylome Signature with Clinicopathologic Parameters and Outcome
2.6. Identification of a Specific Methylome Signature Associated with Metastasis from Acral Melanomas (AMs)
2.7. Association of Aberrantly Methylated Genes in Metastatic AM with Clinicopathologic Parameters and Outcome
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. DNA Isolation, Methylation Analysis, Functional Genomic Pathway Analysis, and Statistical Analysis
4.3. Data Access
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bradford, P.T.; Goldstein, A.M.; McMaster, M.L.; Tucker, M.A. Acral lentiginous melanoma: Incidence and survival patterns in the United States, 1986–2005. Arch. Dermatol. 2009, 145, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Durbec, F.; Martin, L.; Derancourt, C.; Grange, F. Melanoma of the hand and foot: Epidemiological, prognostic and genetic features. A systematic review. Br. J. Dermatol. 2012, 166, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Ugorji, R.; Khachemoune, A. Acral melanoma foot lesions. Part 1: Epidemiology, aetiology, and molecular pathology. Clin. Exp. Dermatol. 2017, 42, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.; Touzet, S.; Dalle, S.; Ronger-Savlé, S.; Balme, B.; Thomas, L. Acral lentiginous melanoma: A clinicoprognostic study of 126 cases. Br. J. Dermatol. 2006, 155, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.; Touzet, S.; Dalle, S.; Ronger-Savlé, S.; Balme, B.; Thomas, L. lentiginous melanoma: Histopathological prognostic features of 121 cases. Br. J. Dermatol. 2007, 157, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.M.; Shen, L.; Sokil, M.M.; Yeh, I.; Jorgenson, E. Prognostic factors and survival in acral lentiginous melanoma. Br. J. Dermatol. 2017, 177, 428–435. [Google Scholar] [CrossRef]
- Cress, R.D.; Holly, E.A. Incidence of cutaneous melanoma among non-Hispanic whites, Hispanics, Asians, and blacks: An analysis of California cancer registry data, 1988–1993. Cancer Causes Control. 1997, 8, 246–252. [Google Scholar] [CrossRef]
- Chang, J.W.; Yeh, K.Y.; Wang, C.H.; Yang, T.S.; Chiang, H.F.; Wei, F.C.; Kuo, T.T.; Yang, C.H. Malignant melanoma in Taiwan: A prognostic study of 181 cases. Melanoma Res. 2004, 14, 537–541. [Google Scholar] [CrossRef]
- Hudson, D.A.; Krige, J.E. Melanoma in black South Africans. J. Am. Coll. Surg. 1995, 180, 65–71. [Google Scholar]
- Niu, H.T.; Zhou, Q.M.; Wang, F.; Shao, Q.; Guan, Y.X.; Wen, X.Z.; Chen, L.Z.; Feng, Q.S.; Li, W.; Zeng, Y.X.; et al. Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment Cell Melanoma Res. 2013, 26, 646–653. [Google Scholar] [CrossRef]
- Turner, J.; Couts, K.; Sheren, J.; Saichaemchan, S.; Ariyawutyakorn, W.; Avolio, I.; Cabral, E.; Glogowska, M.; Amato, C.; Robinson, S.; et al. Kinase gene fusions in defined subsets of melanoma. Pigment Cell Melanoma Res. 2017, 30, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, I.; Jorgenson, E.; Shen, L.; Xu, M.; North, J.P.; Shain, A.H.; Reuss, D.; Wu, H.; Robinson, W.A.; Olshen, A.; et al. Targeted genomic profiling of acral melanoma. J. Natl. Cancer Inst. 2019, 111, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 239–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinke, C.; Mo, Y.; Yu, Y.; Amiri, K.; Sosman, J.; Greally, J.; Verma, A. Aberrant DNA methylation in malignant melanoma. Melanoma Res. 2010, 20, 253–265. [Google Scholar] [CrossRef]
- Ecsedi, S.; Hernandez-Vargas, H.; Lima, S.C.; Vizkeleti, L.; Toth, R.; Lazar, V.; Koroknai, V.; Kiss, T.; Emri, G.; Herceg, Z.; et al. DNA methylation characteristics of primary melanomas with distinct biological behaviour. PLoS ONE 2014, 9, e96612. [Google Scholar] [CrossRef] [Green Version]
- Thomas, N.E.; Slater, N.A.; Edmiston, S.N.; Zhou, X.; Kuan, P.F.; Groben, P.A.; Carson, C.C.; Hao, H.; Parrish, E.; Moschos, S.J.; et al. DNA methylation profiles in primary cutaneous melanomas are associated with clinically significant pathologic features. Pigment Cell Melanoma Res. 2014, 27, 1097–1105. [Google Scholar] [CrossRef]
- Hoon, D.S.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004, 23, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Roh, M.R.; Gupta, S.; Park, K.H.; Chung, K.Y.; Lauss, M.; Flaherty, K.T.; Jönsson, G.; Rha, S.Y.; Tsao, H. Promoter methylation of PTEN is a significant prognostic factor in melanoma survival. J. Investig. Dermatol. 2016, 136, 1002–1011. [Google Scholar] [CrossRef]
- De Unamuno Bustos, B.; Murria Estal, R.; Pérez Simó, G.; Simarro Farinos, J.; Pujol Marco, C.; Navarro Mira, M.; Alegre de Miquel, V.; Ballester Sánchez, R.; Sabater Marco, V.; Llavador Ros, M.; et al. Aberrant DNA methylation is associated with aggressive clinicopathological features and poor survival in cutaneous melanoma. Br. J. Dermatol. 2018, 179, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, S.; Fiorentini, G.; Clementi, M.; Palumbo, G.; Masedu, F.; Deraco, M.; De Manzoni, G.; Chiominto, A.; Valenti, M.; Pellegrini, C. MGMT methylation correlates with melphalan pelvic perfusion survival in stage III melanoma patients: A pilot study. Melanoma Res. 2017, 27, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigalotti, L.; Covre, A.; Fratta, E.; Parisi, G.; Sonego, P.; Colizzi, F.; Coral, S.; Massarut, S.; Kirkwood, J.M.; Maio, M. Whole genome methylation profiles as independent markers of survival in stage IIIC melanoma patients. J. Transl. Med. 2012, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; O’Day, S.J.; Umetani, N.; Martinez, S.R.; Kitago, M.; Koyanagi, K.; Kuo, C.; Takeshima, T.L.; Milford, R.; Wang, H.J.; et al. Predictive utility of circulating methylated DNA in serum of melanoma patients receiving biochemotherapy. J. Clin. Oncol. 2005, 23, 9351–9358. [Google Scholar] [CrossRef]
- Mori, T.; Martinez, S.R.; O’Day, S.J.; Morton, D.L.; Umetani, N.; Kitago, M.; Tanemura, A.; Nguyen, S.L.; Tran, A.N.; Wang, H.J.; et al. Estrogen receptor-alpha methylation predicts melanoma progression. Cancer Res. 2006, 66, 6692–6698. [Google Scholar] [CrossRef] [Green Version]
- Marzese, D.M.; Scolyer, R.A.; Huynh, J.L.; Huang, S.K.; Hirose, H.; Chong, K.K.; Kiyohara, E.; Wang, J.; Kawas, N.P.; Donovan, N.C.; et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum. Mol. Genet. 2014, 23, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Gambichler, T.; Sand, M.; Skrygan, M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res. 2013, 23, 218–220. [Google Scholar] [CrossRef]
- Gauvrit, S.; Villasenor, A.; Strilic, B.; Kitchen, P.; Collins, M.M.; Marín-Juez, R.; Guenther, S.; Maischein, H.M.; Fukuda, N.; Canham, M.A.; et al. HHEX is a transcriptional regulator of the VEGFC/FLT4/PROX1 signaling axis during vascular development. Nat. Commun. 2018, 9, 2704. [Google Scholar] [CrossRef] [Green Version]
- Goodings, C.; Smith, E.; Mathias, E.; Elliott, N.; Cleveland, S.M.; Tripathi, R.M.; Layer, J.H.; Chen, X.; Guo, Y.; Shyr, Y.; et al. Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells 2015, 33, 2628–2641. [Google Scholar] [CrossRef] [Green Version]
- George, A.; Morse, H.C., 3rd; Justice, M.J. The homeobox gene Hex induces T-cell-derived lymphomas when overexpressed in hematopoietic precursor cells. Oncogene 2003, 22, 6764–6773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Ma, L.; Chen, Y.; Xu, H.; Chen, S.; Wu, W. Overexpression of HHEX in acute myeloid leukemia with t(8;21)(q22;q22) translocation. Ann. Clin. Lab. Sci. 2017, 47, 687–697. [Google Scholar] [PubMed]
- Parada, H., Jr.; Cleveland, R.J.; North, K.E.; Stevens, J.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M.; Martinez, M.E.; Gammon, M.D. Genetic polymorphisms of diabetes-related genes, their interaction with diabetes status, and breast cancer incidence and mortality: The Long Island Breast Cancer Study Project. Mol. Carcinog. 2019, 58, 436–446. [Google Scholar] [PubMed]
- Kershaw, R.M.; Siddiqui, Y.H.; Roberts, D.; Jayaraman, P.S.; Gaston, K. PRH/HHex inhibits the migration of breast and prostate epithelial cells through direct transcriptional regulation of Endoglin. Oncogene 2014, 33, 5592–5600. [Google Scholar] [CrossRef] [Green Version]
- Gaston, K.; Tsitsilianos, M.A.; Wadey, K.; Jayaraman, P.S. Misregulation of the proline rich homeodomain (PRH/HHEX) protein in cancer cells and its consequences for tumour growth and invasion. Cell Biosci. 2016, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Liu, J.P.; Gao, C.; Xiong, Y.Y.; Li, M.; Wang, Y.P.; Su, Y.W.; Lin, M.; Jiang, A.L.; Xiong, L.F.; et al. Two variants on T2DM susceptible gene HHEX are associated with CRC risk in a Chinese population. Oncotarget 2016, 7, 29770–29779. [Google Scholar] [CrossRef]
- Siddiqui, Y.H.; Kershaw, R.M.; Humphreys, E.H.; Assis Junior, E.M.; Chaudhri, S.; Jayaraman, P.S.; Gaston, K. CK2 abrogates the inhibitory effects of PRH/HHEX on prostate cancer cell migration and invasion and acts through PRH to control cell proliferation. Oncogenesis 2017, 6, e293. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; You, P.; Zhao, J.P.; Zhang, S.L.; Song, S.H.; Fu, Z.R.; Ye, L.W.; Zi, X.Y.; Xie, D.F.; Zhu, M.H.; et al. A potential role for the homeoprotein Hhex in hepatocellular carcinoma progression. Med. Oncol. 2012, 29, 1059–1067. [Google Scholar] [CrossRef]
- Yun, H.; Bedolla, R.; Horning, A.; Li, R.; Chiang, H.C.; Huang, T.H.; Reddick, R.; Olumi, A.F.; Ghosh, R.; Kumar, A.P. BRCA1 interacting protein COBRA1 facilitates adaptation to castrate-resistant growth conditions. Int. J. Mol. Sci. 2018, 19, 2104. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Song, S.; Jiang, Z.; Yan, J.; Lu, Q.; Huang, C.; Ye, Q. Characterization of COBRA1 in human breast cancer cell lines using a new polyclonal antibody against COBRA1. IUBMB Life 2004, 56, 161–166. [Google Scholar] [CrossRef]
- McChesney, P.A.; Aiyar, S.E.; Lee, O.J.; Zaika, A.; Moskaluk, C.; Li, R.; El-Rifai, W. Cofactor of BRCA1: A novel transcription factor regulator in upper gastrointestinal adenocarcinomas. Cancer Res. 2006, 66, 1346–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nallet-Staub, F.; Marsaud, V.; Li, L.; Gilbert, C.; Dodier, S.; Bataille, V.; Sudol, M.; Herlyn, M.; Mauviel, A. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J. Investig. Dermatol. 2014, 134, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigi, F.; Schmeckpeper, J.; Pow-Anpongkul, P.; Payne, J.A.; Zhang, L.; Zhang, Z.; Huang, J.; Mirotsou, M.; Dzau, V.J. C3orf58, a novel paracrine protein, stimulates cardiomyocyte cell-cycle progression through the PI3K-AKT-CDK7 pathway. Circ. Res. 2013, 113, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogony, J.; Choi, H.J.; Lui, A.; Cristofanilli, M.; Lewis-Wambi, J. Interferon-induced transmembrane protein 1 (IFITM1) overexpression enhances the aggressive phenotype of SUM149 inflammatory breast cancer cells in a signal transducer and activator of transcription 2 (STAT2)-dependent manner. Breast Cancer Res. 2016, 18, 25. [Google Scholar] [CrossRef] [Green Version]
- Hatano, H.; Kudo, Y.; Ogawa, I.; Tsunematsu, T.; Kikuchi, A.; Abiko, Y.; Takata, T. IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin. Cancer Res. 2008, 14, 6097–6105. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Xie, D.; Ng, S.S.; Lum, C.T.; Cai, M.Y.; Cheung, W.K.; Kung, H.F.; Lin, G.; Wang, X.; Lin, M.C. IFITM1 promotes the metastasis of human colorectal cancer via CAV-1. Cancer Lett. 2015, 368, 135–143. [Google Scholar] [CrossRef]
- Lui, A.J.; Geanes, E.S.; Ogony, J.; Behbod, F.; Marquess, J.; Valdez, K.; Jewell, W.; Tawfik, O.; Lewis-Wambi, J. IFITM1 suppression blocks proliferation and invasion of aromatase inhibitor-resistant breast cancer in vivo by JAK/STAT-mediated induction of p21. Cancer Lett. 2017, 399, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhao, Z.; Yi, X.; Zuo, Q.; Li, H.; Guo, X.; Li, D.; He, H.; Pan, Z.; Fan, P.; et al. Down-regulation of IFITM1 and its growth inhibitory role in cervical squamous cell carcinoma. Cancer Cell Int. 2017, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Amara, S.; Majors, C.; Roy, B.; Hill, S.; Rose, K.L.; Myles, E.L.; Tiriveedhi, V. Critical role of SIK3 in mediating high salt and IL-17 synergy leading to breast cancer cell proliferation. PLoS ONE 2017, 12, e0180097. [Google Scholar] [CrossRef]
- Du, W.Q.; Zheng, J.N.; Pei, D.S. The diverse oncogenic and tumor suppressor roles of salt-inducible kinase (SIK) in cancer. Expert Opin. Ther. Targets 2016, 20, 477–485. [Google Scholar] [CrossRef]
- Walkinshaw, D.R.; Weist, R.; Kim, G.W.; You, L.; Xiao, L.; Nie, J.; Li, C.S.; Zhao, S.; Xu, M.; Yang, X.J. The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 2013, 288, 9345–9362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.T.; Asokan, S.B.; King, S.J.; Bo, T.; Dubose, E.S.; Liu, W.; Berginski, M.E.; Simon, J.M.; Davis, I.J.; Gomez, S.M.; et al. LKB1 loss in melanoma disrupts directional migration toward extracellular matrix cues. J. Cell Biol. 2014, 207, 299–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jour, G.; Vasudevaraja, V.; Prieto, V.G.; Snuderl, M.; Torres-Cabala, C.A.; Al-Rohil, R.; Sulman, E.P.; Ballester, L.Y.; Aung, P.P. BCAT1 and miR-2504: Novel methylome signature distinguishes spindle/desmoplastic melanoma from superficial malignant peripheral nerve sheath tumor. Mod. Pathol. 2019, 32, 338–345. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | All (N = 48) | PALM (N = 22) | MALM (N = 9) † | NALM (N = 8) † | PCM (N = 9) | p Value # |
|---|---|---|---|---|---|---|
| Sex | ||||||
| Male | 30 (63) | 14 (64) | 5 (56) | 5 (63) | 6 (67) | 0.97 |
| Female | 18 (38) | 8 (36) | 4 (44) | 3 (38) | 3 (33) | |
| Age, median (range), y | 62.1 (1.7–90.6) | 67.4 (36.6–90.6) | 58.4 (16.0–79.0) | 55.6 (1.7–89.4) | 62.3 (13.7–79.0) | 0.22 |
| Race | ||||||
| White | 40 (83) | 16 (73) | 8 (89) | 7 (88) | 9 (100) | 0.34 |
| Other | 8 (17) | 6 (27) | 1 (11) | 1 (13) | 0 | |
| AJCC 8th edition stage at presentation | ||||||
| I | 10 (21) | 6 (27) | 1 (13) | 1 (13) | 2 (22) | 0.37 |
| II | 15 (32) | 7 (32) | 1 (13) | 2 (25) | 5 (56) | |
| III | 21 (45) | 9 (41) | 6 (75) | 4 (50) | 2 (22) | |
| IV | 1 (2) | 0 | 0 | 1 (13) | 0 | |
| Site(s) of LN metastasis at diagnosis | ||||||
| None | 25 (54) | 13 (59) | 3 (33) | 1 (17) | 8 (89) | 0.019 |
| Enlarged ‡ | 1 (2) | 1 (5) | 0 | 0 | 0 | |
| Regional | 1 (2) | 0 | 1 (11) | 0 | 0 | |
| Sentinel | 17 (37) | 8 (36) | 3 (33) | 5 (83) | 1 (11) | |
| Regional and sentinel | 2 (4) | 0 | 2 (22) | 0 | 0 | |
| Total number of LNs with metastasis at diagnosis, median (range) | 2 (1–11) | 2 (1–3) | 4 (1–11) | 2 (1–2) | 1 (1–1) | 0.24 |
| Primary histologic subtype | ||||||
| ALM | 31 (65) | 22 (100) | 9 (100) | 0 | 0 | <0.001 |
| LMM | 2 (4) | 0 | 0 | 0 | 2 (22) | |
| NM | 6 (13) | 0 | 0 | 4 (50) | 2 (22) | |
| SSM | 5 (10) | 0 | 0 | 3 (38) | 2 (22) | |
| Unclassified | 4 (8) | 0 | 0 | 1 (13) | 3 (33) | |
| Clark level | ||||||
| II | 1 (2) | 1 (5) | 0 | 0 | 0 | 0.79 |
| III | 1 (2) | 0 | 0 | 1 (13) | 0 | |
| IV | 32 (68) | 15 (68) | 6 (75) | 4 (50) | 7 (78) | |
| V | 13 (27) | 6 (27) | 2 (22) | 3 (38) | 2 (22) | |
| Breslow thickness, mm | ||||||
| 1.01–2 | 13 (28) | 7 (33) | 2 (25) | 2 (25) | 2 (22) | 0.34 |
| 2.01–4 | 14 (30) | 4 (19) | 1 (13) | 4 (50) | 5 (56) | |
| >4 | 19 (41) | 10 (48) | 5 (63) | 2 (25) | 2 (22) | |
| Radial growth phase | ||||||
| Present | 25 (74) | 13 (87) | 6 (100) | 2 (33) | 4 (57) | 0.021 |
| Not identified | 9 (26) | 2 (13) | 0 | 4 (67) | 3 (43) | |
| Vertical growth phase | ||||||
| Present | 46 (100) | 22 (100) | 8 (100) | 7 (100) | 9 (100) | |
| Not identified | 0 | 0 | 0 | 0 | 0 | |
| Mitotic figures | ||||||
| <1 | 2 (5) | 1 (5) | 0 | 1 (17) | 0 | 0.49 |
| 1–4 | 30 (68) | 14 (64) | 5 (63) | 3 (50) | 8 (100) | |
| 5–9 | 5 (11) | 3 (14) | 2 (25) | 0 | 0 | |
| 10–20 | 5 (11) | 2 (9) | 1 (13) | 2 (33) | 0 | |
| >20 | 2 (5) | 2 (9) | 0 | 0 | 0 | |
| Ulceration | ||||||
| Present | 21 (48) | 11 (52) | 6 (75) | 2 (29) | 2 (25) | 0.18 |
| Not identified | 23 (52) | 10 (48) | 2 (25) | 5 (71) | 6 (75) | |
| Regression | ||||||
| Present | 6 (14) | 4 (18) | 1 (13) | 0 | 1 (13) | 0.92 |
| Not identified | 38 (86) | 18 (82) | 7 (88) | 6 (100) | 7 (88) | |
| Vascular invasion | ||||||
| Present | 6 (13) | 2 (9) | 1 (11) | 3 (50) | 0 | 0.05 |
| Not identified | 40 (87) | 20 (91) | 8 (89) | 3 (50) | 9 (100) | |
| Perineural invasion | ||||||
| Present | 13 (30) | 7 (32) | 3 (38) | 2 (33) | 1 (13) | 0.73 |
| Not identified | 31 (70) | 15 (68) | 5 (63) | 4 (67) | 7 (88) | |
| Microscopic satellitosis | ||||||
| Present | 4 (9) | 2 (9) | 0 | 1 (17) | 1 (13) | 0.71 |
| Not identified | 40 (91) | 20 (91) | 8 (100) | 5 (83) | 7 (88) | |
| TIL | ||||||
| Non-brisk | 45 (100) | 22 (100) | 8 (100) | 7 (100) | 8 (100) | |
| Brisk | 0 | 0 | 0 | 0 | 0 | |
| Associated nevus | ||||||
| Present | 3 (7) | 1 (5) | 0 | 0 | 2 (25) | 0.23 |
| Not identified | 41 (93) | 21 (95) | 8 (100) | 6 (100) | 6 (75) | |
| Predominant cytology | ||||||
| Epithelioid | 28 (64) | 14 (67) | 4 (50) | 6 (86) | 4 (50) | 0.22 |
| Nevoid | 5 (11) | 2 (10) | 0 | 0 | 3 (38) | |
| Spindled | 11 (25) | 5 (24) | 4 (50) | 1 (14) | 1 (13) | |
| Genetic mutation | ||||||
| Yes | 15 (48) | 6 (38) | 4 (44) | 3 (100) | 2 (67) | 0.29 |
| No | 16 (52) | 10 (63) | 5 (56) | 0 | 1 (33) | |
| Vital status | ||||||
| Alive with NED | 20 (42) | 12 (55) | 1 (11) | 2 (25) | 5 (56) | 0.006 |
| Alive with disease | 7 (15) | 5 (23) | 2 (22) | 0 | 0 | |
| Died of other causes | 8 (17) | 3 (14) | 0 | 2 (25) | 3 (33) | |
| Died with disease | 13 (27) | 2 (9) | 6 (67) | 4 (50) | 1 (11) | |
| Follow-up time after diagnosis (all patients), median (range), mo | 54.9 (5.2–186.0) | 47.7 (11.0–186.0) | 72.1 (25.0–141.1) | 43.0 (7.1–147.8) | 93.7 (5.2–123.9) | 0.42 |
| Survival time after diagnosis (survivors) §, months | ||||||
| Number of patients | 27 | 17 | 3 | 2 | 5 | 0.07 |
| Median (range) | 68.0 (5.2–186.0) | 48.9 (18.6–186.0) | 103.8 (29.7–141.1) | 136.5 (125.1–147.8) | 93.7 (57.6–123.9) |
| Survival | Genes | HR (95% CI) | p Value |
| Overall | CDH13 | 0.29 (0.10 to 0.84) | 0.022 |
| NELFB | 4.75 (1.61 to 13.99) | 0.005 | |
| HHEX | 4.60 (1.63 to 13.00) | 0.004 | |
| Disease-specific | CDH13 | 0.22 (0.06 to 0.74) | 0.014 |
| DIPK2A | 5.09 (1.11 to 23.34) | 0.036 | |
| NELFB | 7.17 (1.92 to 26.76) | 0.003 | |
| TEF | 4.59 (1.21 to 17.43) | 0.025 | |
| HHEX | 6.13 (1.83 to 20.49) | 0.003 | |
| SIK3 | 3.26 (1.02 to 10.39) | 0.046 | |
| IFITM1 | 3.88 (1.04 to 14.48) | 0.044 | |
| Histologic Parameter | Genes | HR (95% CI) | p-value |
| Lymph node metastasis at diagnosis (yes vs. no) | CDH13 | 0.14 (0.02 to 0.77) | 0.025 |
| NELFB | 5.50 (1.16 to 26.14) | 0.032 | |
| HHEX | 7.50 (1.61 to 34.95) | 0.010 | |
| Predominant histologic subtype (ALM vs. other) | DIPK2A | 0.04 (0.00 to 0.38) | 0.005 |
| NELFB | 0.04 (0.00 to 0.93) | 0.044 | |
| TEF | 0.06 (0.01 to 0.38) | 0.003 | |
| HHEX | 0.06 (0.01 to 0.38) | 0.003 | |
| IFITM1 | 10.82 (1.17 to 100.4) | 0.036 | |
| Breslow thickness (>4 mm vs. 1.01–4 mm) | CDH13 | 0.12 (0.02 to 0.59) | 0.009 |
| Mitotic figures (≥10 vs. <4) | CDH13 | 0.19 (0.04 to 0.92) | 0.040 |
| TEF | 14.44 (1.56 to 133.6) | 0.019 | |
| SIK3 | 9.07 (1.72 to 47.67) | 0.009 | |
| Ulceration (present vs. not identified) | CDH13 | 0.06 (0.01 to 0.56) | 0.013 |
| SIK3 | 7.94 (1.60 to 39.42) | 0.011 | |
| PNI (present vs. not identified) | CDH13 | 0.07 (0.01 to 0.64) | 0.019 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pradhan, D.; Jour, G.; Milton, D.; Vasudevaraja, V.; Tetzlaff, M.T.; Nagarajan, P.; Curry, J.L.; Ivan, D.; Long, L.; Ding, Y.; et al. Aberrant DNA Methylation Predicts Melanoma-Specific Survival in Patients with Acral Melanoma. Cancers 2019, 11, 2031. https://doi.org/10.3390/cancers11122031
Pradhan D, Jour G, Milton D, Vasudevaraja V, Tetzlaff MT, Nagarajan P, Curry JL, Ivan D, Long L, Ding Y, et al. Aberrant DNA Methylation Predicts Melanoma-Specific Survival in Patients with Acral Melanoma. Cancers. 2019; 11(12):2031. https://doi.org/10.3390/cancers11122031
Chicago/Turabian StylePradhan, Dinesh, George Jour, Denái Milton, Varshini Vasudevaraja, Michael T. Tetzlaff, Priyadharsini Nagarajan, Jonathan L. Curry, Doina Ivan, Lihong Long, Yingwen Ding, and et al. 2019. "Aberrant DNA Methylation Predicts Melanoma-Specific Survival in Patients with Acral Melanoma" Cancers 11, no. 12: 2031. https://doi.org/10.3390/cancers11122031