Discovering Rare Genes Contributing to Cancer Stemness and Invasive Potential by GBM Single-Cell Transcriptional Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
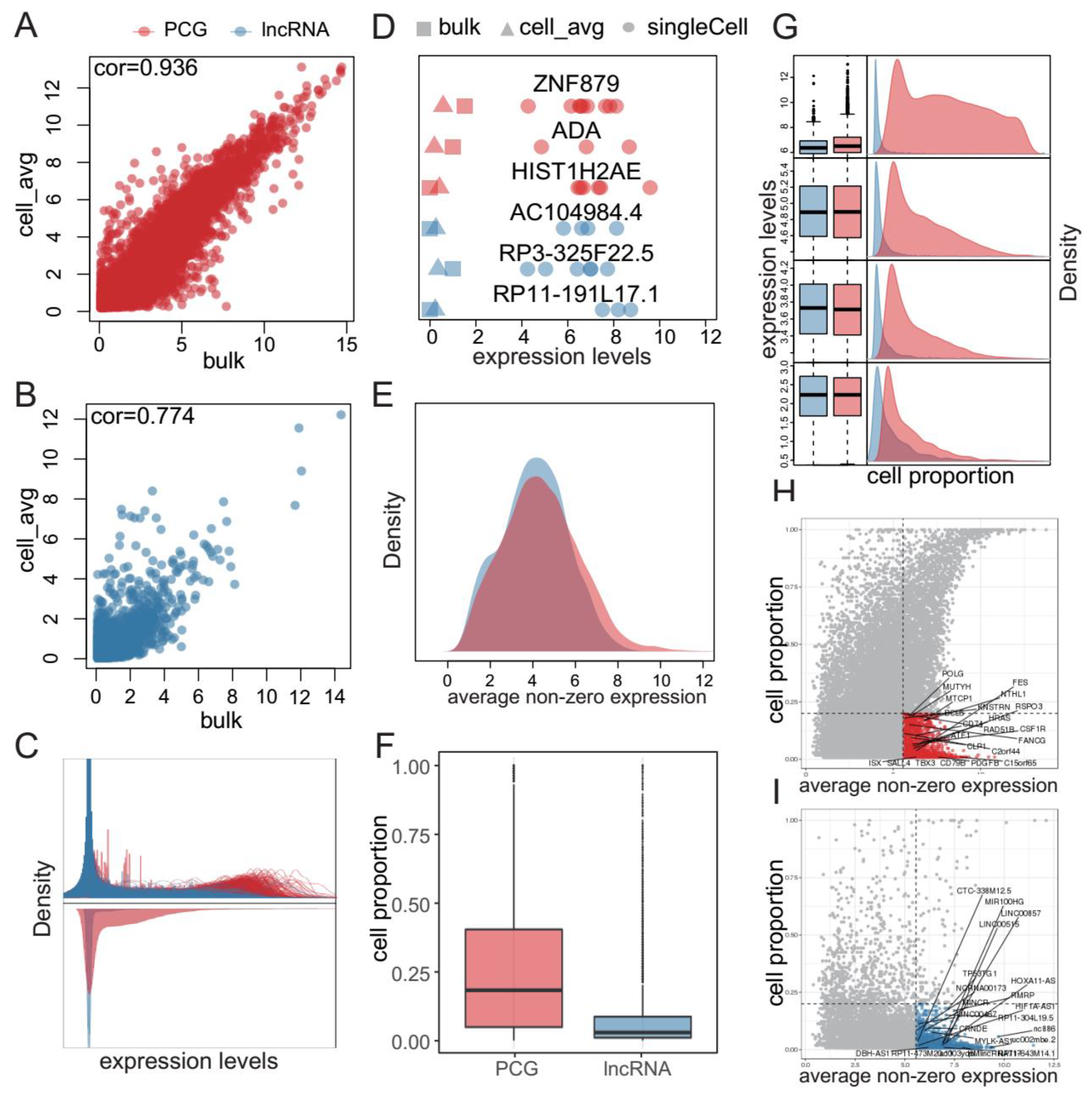
2.1. Single-Cell Profiling Uncovers Rare Genes in GBM
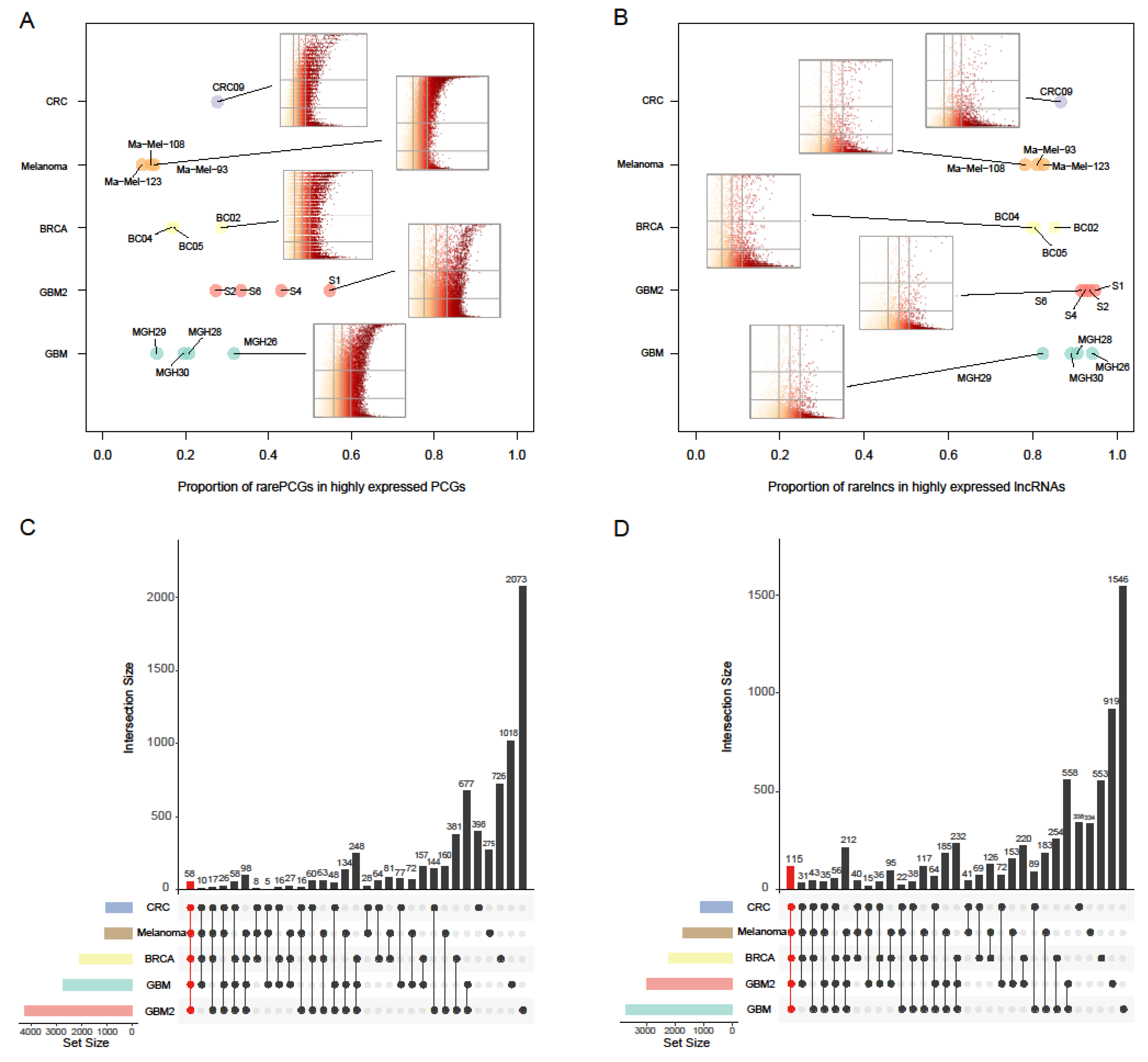
2.2. Rare PCGs/lncRNAs Present Extensively in Various Cancers
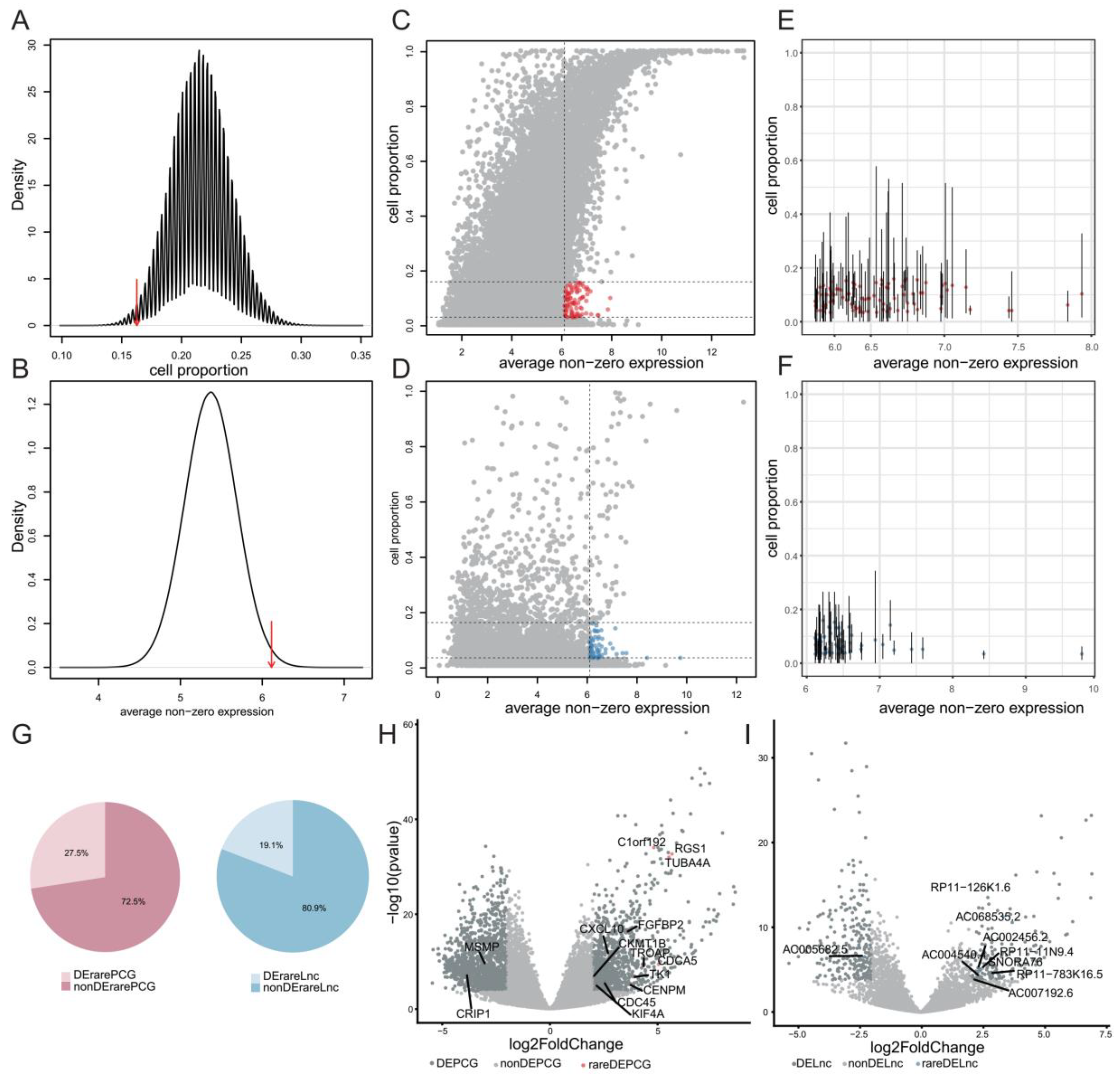
2.3. Systematical Identification of Rare Genes in GBM
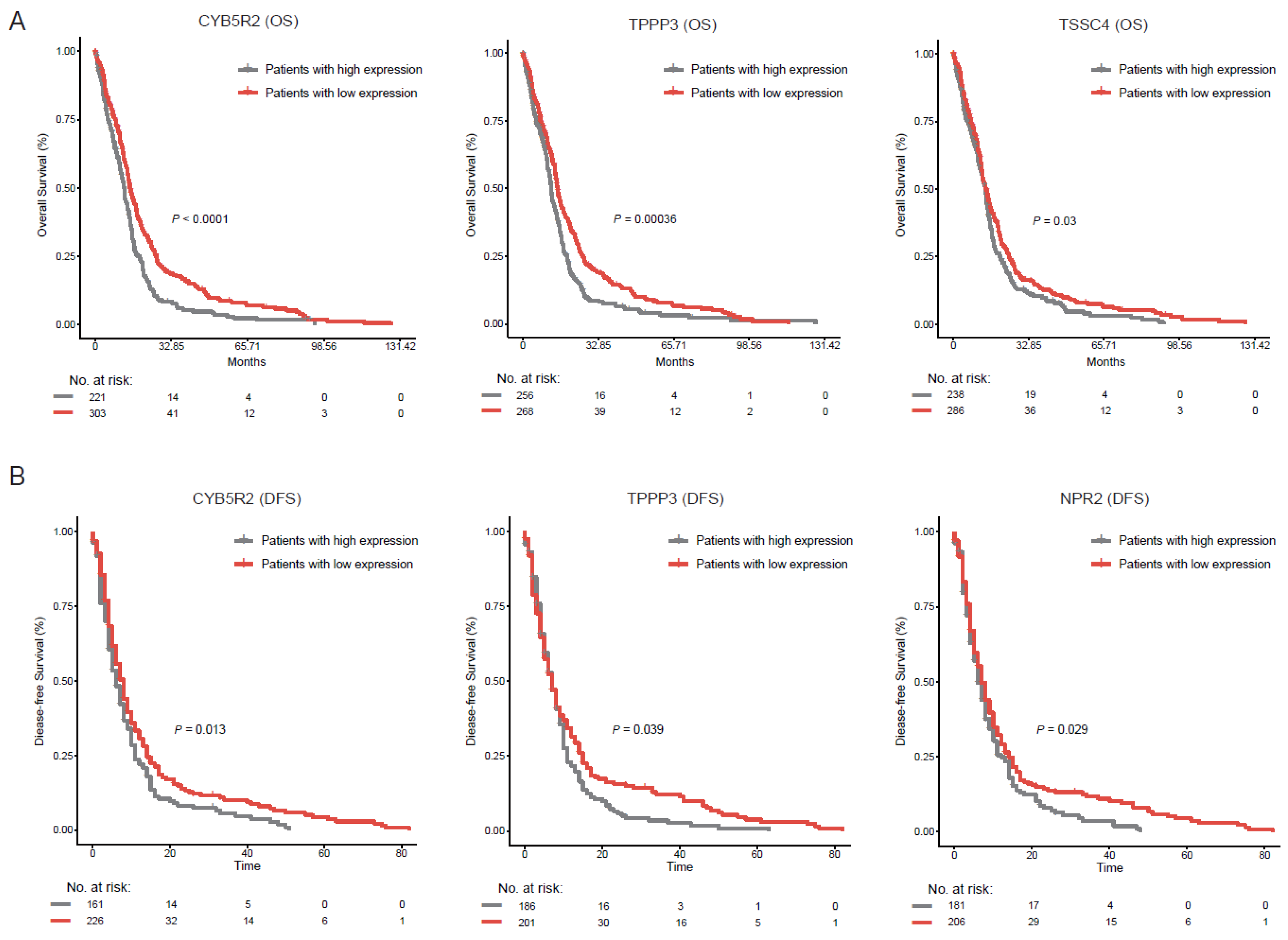
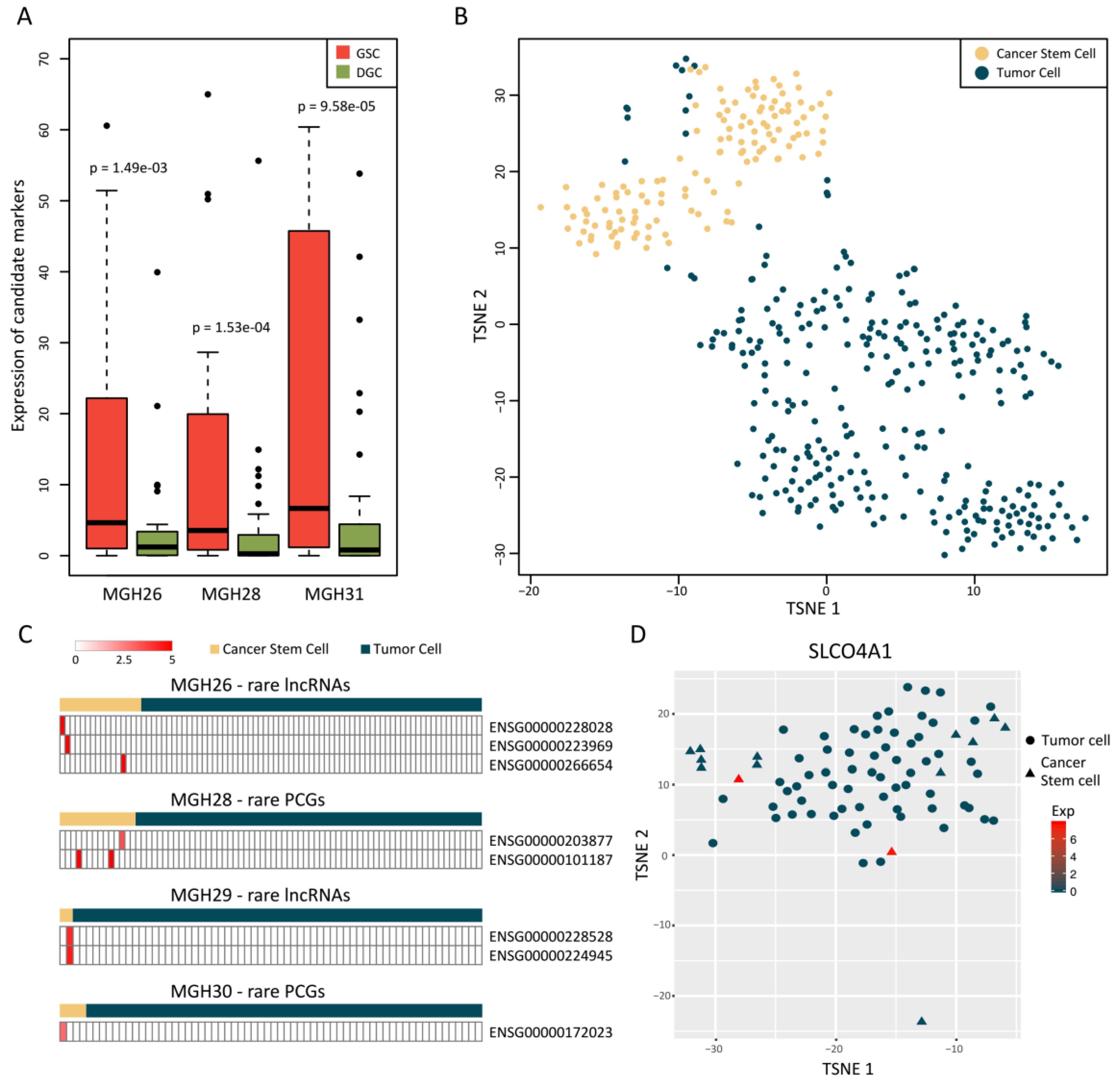
2.4. Rare Genes Are Related with Cancer Stemness of GBM
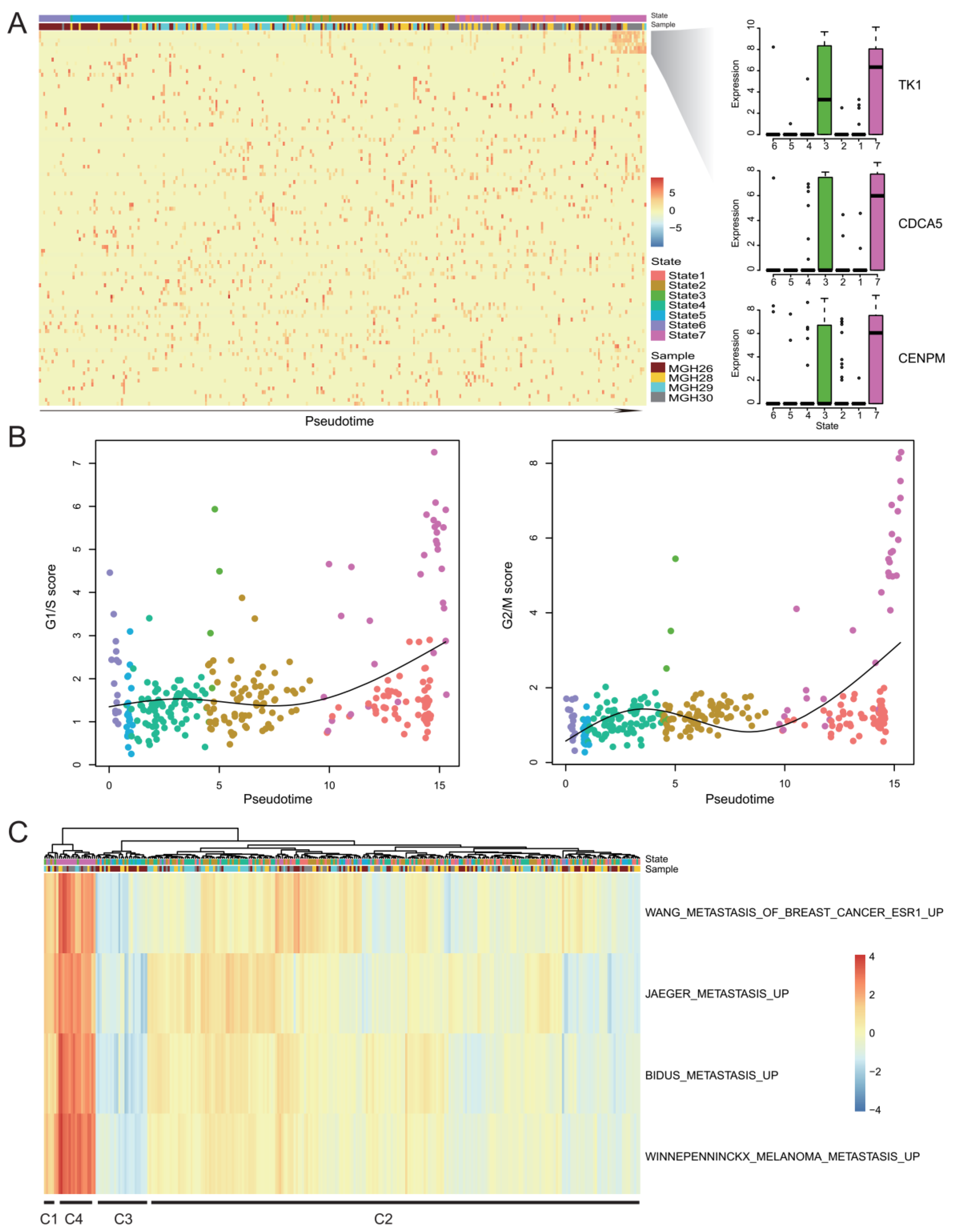
2.5. Rare Genes Reveal a Subpopulation of Cancer Cell with High Proliferation Activity and Invasive Potential
3. Discussion
4. Materials and Methods
4.1. Processing of Publicly Available Single-Cell RNA-Seq Data of GBM
4.2. Removing Potential Non-Tumor Cells
4.3. Filtering Expression Noise for lncRNAs Based on a Classification Model
4.4. Identifying Rare Genes in GBM
4.5. Differential Analysis and Survival Analysis on GBM Tissue Samples from TCGA
4.6. Single Cell Trajectory Analysis
4.7. Cell Cycle Analysis
4.8. Single Cell Differential Expression Analysis
4.9. Invasive Potential Score (IPS)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, I.C.; Svensson, V.; Labalette, C.; Ferreira, L.; Hamey, F.; Voet, T.; Teichmann, S.A.; Cvejic, A. Single-Cell RNA-Sequencing Reveals a Continuous Spectrum of Differentiation in Hematopoietic Cells. Cell Rep. 2016, 14, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Deng, Q.; Ramskold, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Munoz-Manchado, A.B.; Codeluppi, S.; Lonnerberg, P.; La Manno, G.; Jureus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Torre, E.; Dueck, H.; Shaffer, S.; Gospocic, J.; Gupte, R.; Bonasio, R.; Kim, J.; Murray, J.; Raj, A. Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH. Cell Syst. 2018, 6, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Lymbouridou, R.; Soufla, G.; Chatzinikola, A.M.; Vakis, A.; Spandidos, D.A. Down-regulation of K-ras and H-ras in human brain gliomas. Eur. J. Cancer 2009, 45, 1294–1303. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Li, J.; Zhang, Y.; Yin, H.; Han, B. CRNDE, a long-noncoding RNA, promotes glioma cell growth and invasion through mTOR signaling. Cancer Lett. 2015, 367, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: A manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016, 44, D980–D985. [Google Scholar] [CrossRef]
- Itou, J.; Tanaka, S.; Li, W.; Iida, A.; Sehara-Fujisawa, A.; Sato, F.; Toi, M. The Sal-like 4 - integrin alpha6beta1 network promotes cell migration for metastasis via activation of focal adhesion dynamics in basal-like breast cancer cells. Biochim. Et Biophys. Acta 2017, 1864, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [Green Version]
- Pinto, J.P.; Kalathur, R.K.; Oliveira, D.V.; Barata, T.; Machado, R.S.; Machado, S.; Pacheco-Leyva, I.; Duarte, I.; Futschik, M.E. StemChecker: A web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015, 43, W72–W77. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, G.; Rusch, M.; Lukes, L.; Buetow, K.H.; Zhang, J.; Hunter, K.W. Integrated cross-species transcriptional network analysis of metastatic susceptibility. Proc. Natl. Acad. Sci. USA 2012, 109, 3184–3189. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, X.; Wang, L.; Zheng, G.; Du, L.; Yang, Y.; Dong, Z.; Liu, Y.; Qu, A.; Wang, C. Investigation of cell free BIRC5 mRNA as a serum diagnostic and prognostic biomarker for colorectal cancer. J. Surg. Oncol. 2014, 109, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, J.; Zhao, J.; Bai, J.H. Proliferation and invasion of colon cancer cells are suppressed by knockdown of TOP2A. J. Cell. Biochem. 2018, 119, 7256–7263. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.J.; Li, J.D.; Zhang, L.; Wang, J.H.; Wan, T.; Zhou, Y.; Tu, H.; Yun, J.P.; Luo, R.Z.; Jia, W.H.; et al. SPAG5 upregulation predicts poor prognosis in cervical cancer patients and alters sensitivity to taxol treatment via the mTOR signaling pathway. Cell Death Dis. 2014, 5, e1247. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Baruah, P.; Manfredi, A.A.; Bianchi, M.E.; Rovere-Querini, P. HMGB1: Guiding immunity from within. Trends Immunol. 2005, 26, 381–387. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. Embo Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Park, Y.M.; Hwang, S.J.; Masuda, K.; Choi, K.M.; Jeong, M.R.; Nam, D.H.; Gorospe, M.; Kim, H.H. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol. Cell. Biol. 2012, 32, 4237–4244. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Bernards, R.; Weinberg, R.A. A progression puzzle. Nature 2002, 418, 823. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. Leaving home early: Reexamination of the canonical models of tumor progression. Cancer Cell 2008, 14, 283–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, H.; Obradovic, M.M.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Tatlow, P.J.; Piccolo, S.R. A cloud-based workflow to quantify transcript-expression levels in public cancer compendia. Sci. Rep. 2016, 6, 39259. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, L.; Hu, J.; Li, F.; Yuan, H.; Yan, M.; Liao, G.; Xu, L.; Pang, B.; Ping, Y.; Xiao, Y.; et al. Discovering Rare Genes Contributing to Cancer Stemness and Invasive Potential by GBM Single-Cell Transcriptional Analysis. Cancers 2019, 11, 2025. https://doi.org/10.3390/cancers11122025
Pang L, Hu J, Li F, Yuan H, Yan M, Liao G, Xu L, Pang B, Ping Y, Xiao Y, et al. Discovering Rare Genes Contributing to Cancer Stemness and Invasive Potential by GBM Single-Cell Transcriptional Analysis. Cancers. 2019; 11(12):2025. https://doi.org/10.3390/cancers11122025
Chicago/Turabian StylePang, Lin, Jing Hu, Feng Li, Huating Yuan, Min Yan, Gaoming Liao, Liwen Xu, Bo Pang, Yanyan Ping, Yun Xiao, and et al. 2019. "Discovering Rare Genes Contributing to Cancer Stemness and Invasive Potential by GBM Single-Cell Transcriptional Analysis" Cancers 11, no. 12: 2025. https://doi.org/10.3390/cancers11122025