Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies



Abstract
:1. Introduction
2. Evolution of Response Criteria and MRD Techniques
3. Treatment of High-Risk SMM
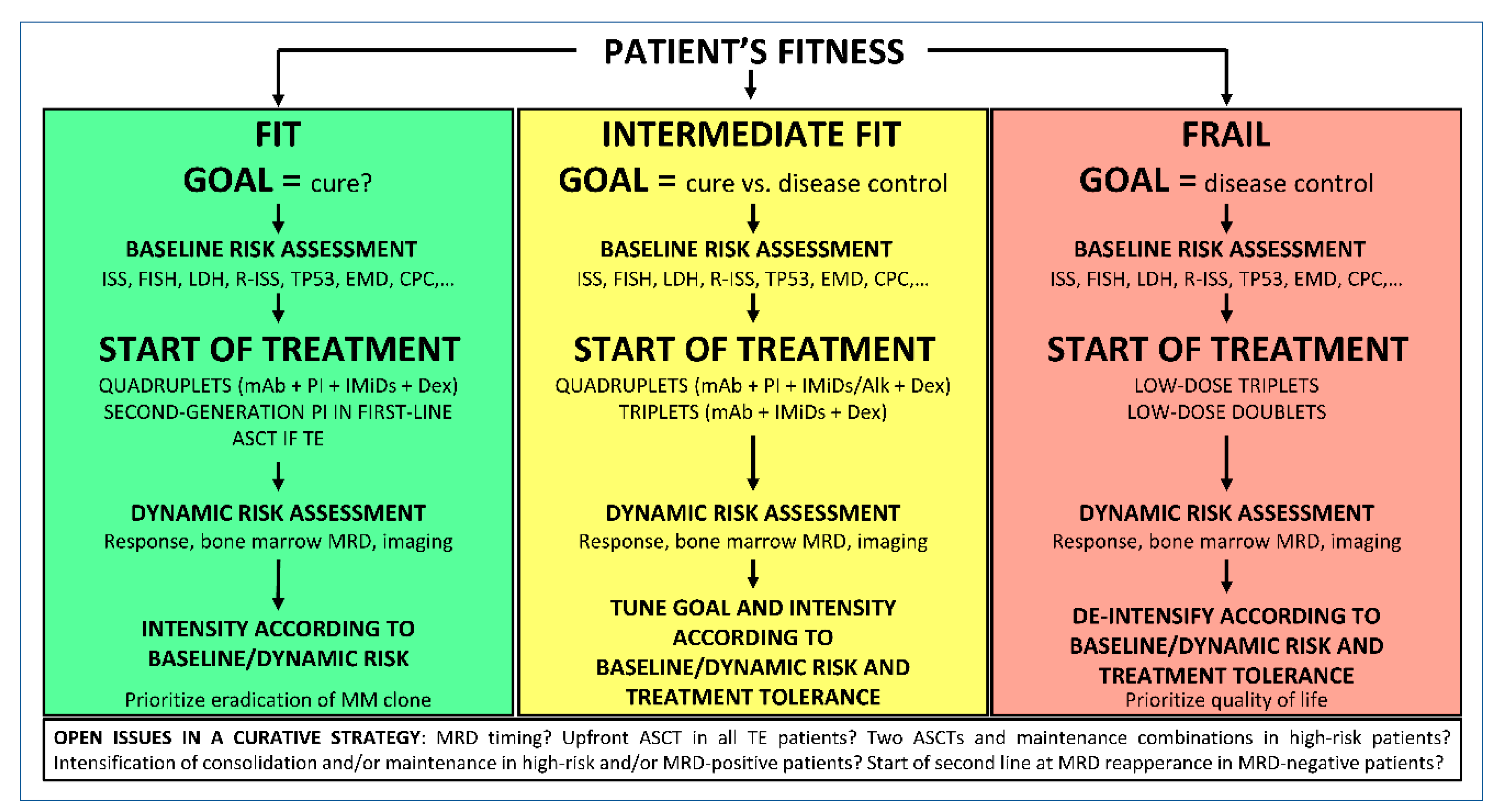
4. Treatment of Symptomatic NDMM
4.1. ASCT-Eligible Patients
4.2. ASCT-Ineligible Patients
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IMWG | International Myeloma Working Group |
| MRD | minimal residual disease |
| NGF | next-generation flow |
| NGS | next-generation sequencing |
| FLC | free light chain |
| M-protein | myeloma protein |
| FCM | flow cytometry |
| SUVmax | maximum standardized uptake value |
| MFC | multiparameter flow cytometry |
| FDG | Fluorodeoxyglucose |
| PET/CT | Positron emission tomography/computed tomography |
| N | number |
| TTP | time to progression |
| y | year |
| NR | not reached |
| CA | chromosomal abnormalities |
| PCs | plasma cells |
| BMPCs | bone marrow PCs |
| HR | hazard ratio |
| FLCr | free light chain ratio |
| FISH | fluorescence in situ hybridization |
| Pts | patients |
| V | bortezomib |
| R, Len | lenalidomide |
| d, dex | dexamethasone |
| T | thalidomide |
| FU | follow-up |
| PFS | progression-free survival |
| OS | overall survival |
| G | grade |
| P | p-value |
| MM | multiple myeloma |
| K | carfilzomib |
| CRAB | hypercalcemia, renal failure, anemia, and bone lesions |
| SMM | smoldering MM |
| ORR | overall response rate |
| Dara | daratumumab |
| ASCT | autologous stem-cell transplantation |
| Y | years |
| NA | not available |
| iv | intravenous |
| D | day |
| AE | adverse event |
| sc | subcutaneous |
| p.o. | orally |
| G-CSF | granulocyte colony-stimulating factor |
| Obs. | observation |
| TE | Transplant-eligible |
| NTE | non-transplant-eligible |
| NDMM | newly diagnosed multiple myeloma |
| C | cyclophosphamide |
| PD | progressive disease |
| MR | minimal response |
| PR | partial response |
| VGPR | very good PR |
| CR | complete response |
| nCR | near CR |
| sCR | stringent CR |
| CHF | congestive heart failure |
| SPMs | second primary malignancies |
| MRD neg | MRD negative/negativity |
| M, Mel | melphalan |
| Mel200 | melphalan at 200 mg/m2 |
| Bu | busulfan |
| p | prednisone |
| Ixa | ixazomib |
| SAEs | serious AEs |
| TEAEs | treatment-emergent AEs |
| GIT | gastrointestinal toxicity |
| QW | given every week |
| Q2W | given every two weeks |
| Q4W | given every 4 weeks |
| CI | confidence interval |
| ISS | International Staging System |
| R-ISS | Revised ISS |
| LDH | lactate dehydrogenase |
| EMD | extramedullary disease |
| CPC | circulating plasma cells |
| mAb | monoclonal antibody |
| PI | proteasome inhibitor |
| IMiDs | immunomodulatory drugs |
References
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, B.; Binder, M.; Dispenzieri, A.; Kapoor, P.; Buadi, F.; Gertz, M.A.; Lacy, M.; Dingli, D.; Hwa, L.; Leung, N.; et al. Continued improvement in survival in multiple myeloma (MM) including high-risk patients. J. Clin. Oncol. 2019, 37. Abstract #8039 [ASCO 2019 Meeting]. [Google Scholar] [CrossRef]
- Castañeda-Avila, M.A.; Ortiz-Ortiz, K.J.; Torres-Cintrón, C.R.; Birmann, B.M.; Epstein, M.M. Trends in cause of death among patients with multiple myeloma in Puerto Rico and the United States SEER population, 1987–2013. Int. J. Cancer 2019, 146, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Delasalle, K.; Feng, L.; Thomas, S.; Giralt, S.; Qazilbash, M.; Handy, B.; Lee, J.J.; Alexanian, R. CR represents an early index of potential long survival in multiple myeloma. Bone Marrow Transplant. 2010, 45, 498–504. [Google Scholar] [CrossRef]
- Ravi, P.; Kumar, S.K.; Cerhan, J.R.; Maurer, M.J.; Dingli, D.; Ansell, S.M.; Rajkumar, S.V. Defining cure in multiple myeloma: A comparative study of outcomes of young individuals with myeloma and curable hematologic malignancies. Blood Cancer J. 2018, 8, 26. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Corre, J.; Garderet, L.; Karlin, L.; Lambert, J.; Macro, M.; et al. Efficacy of daratumumab (DARA) + bortezomib/thalidomide/dexamethasone (D-VTd) in transplant-eligible newly diagnosed multiple myeloma (TE NDMM) based on minimal residual disease (MRD) status: Analysis of the CASSIOPEIA trial. J. Clin. Oncol. 2019, 37. Abstract #8017 [ASCO 2019 International Meeting]. [Google Scholar] [CrossRef]
- Zimmerman, T.; Raje, N.S.; Vij, R.; Reece, D.; Berdeja, J.G.; Stephens, L.A.; McDonnell, K.; Rosenbaum, C.A.; Jasielec, J.; Richardson, P.G.; et al. Final Results of a Phase 2 Trial of Extended Treatment (tx) with Carfilzomib (CFZ), Lenalidomide (LEN), and Dexamethasone (KRd) Plus Autologous Stem Cell Transplantation (ASCT) in Newly Diagnosed Multiple Myeloma (NDMM). Blood 2016, 128. Abstract #675 [ASH 2016 58th Meeting]. [Google Scholar] [CrossRef]
- Gay, F.; Cerrato, C.; Petrucci, M.T.; Zambello, R.; Gamberi, B.; Ballanti, S.; Omedè, P.; Palmieri, S.; Troia, R.; Spada, S.; et al. Efficacy of carfilzomib lenalidomide dexamethasone (KRd) with or without transplantation in newly diagnosed myeloma according to risk status: Results from the forte trial. J. Clin. Oncol. 2019, 37. Abstract #8002 [ASCO 2019 Annual Meeting]. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Rodriguez, C.; Reeves, B.; Nathwani, N.; Costa, L.J.; Lutska, Y.; Hoehn, D.; Pei, H.; Ukropec, J.; Qi, M.; et al. Efficacy and Updated Safety Analysis of a Safety Run-in Cohort from Griffin, a Phase 2 Randomized Study of Daratumumab (Dara), Bortezomib (V), Lenalidomide (R), and Dexamethasone (D.; Dara-Vrd) Vs. Vrd in Patients (Pts) with Newly Diagnosed (ND) Multiple, M. Blood 2018, 132. Abstract #151 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Chari, A.; Lonial, S.; Weiss, B.M.; Comenzo, R.L.; Wu, K.; Khokhar, N.Z.; Wang, J.; Doshi, P.; Usmani, S.Z. Daratumumab (DARA) in combination with carfilzomib, lenalidomide, and dexamethasone (KRd) in patients (pts) with newly diagnosed multiple myeloma (MMY1001): An open-label, phase 1b study. J. Clin. Oncol. 2017, 35. Abstract #8000 [ASCO 2017 Annual Meeting]. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Schmidt-Wolf, I.; Sonneveld, P.; van der Holt, B.; Martin, H.; Barge, R.; Bertsch, U.; Schlenzka, J.; Bos, G.M.J.; Croockewit, S.; et al. Thalidomide in induction treatment increases the very good partial response rate before and after high-dose therapy in previously untreated multiple myeloma. Haematologica 2008, 93, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta, J.-J.; Paiva, B.; Vidriales, M.-B.; Cordón, L.; Cedena, M.-T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.-L.; et al. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V.; Harousseau, J.-L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Paiva, B.; Gutiérrez, N.C.; Rosiñol, L.; Vídriales, M.-B.; Montalbán, M.-Á.; Martínez-López, J.; Mateos, M.-V.; Cibeira, M.-T.; Cordón, L.; Oriol, A.; et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood 2012, 119, 687–691. [Google Scholar] [CrossRef]
- Nanni, C.; Zamagni, E.; Versari, A.; Chauvie, S.; Bianchi, A.; Rensi, M.; Bellò, M.; Rambaldi, I.; Gallamini, A.; Patriarca, F.; et al. Image interpretation criteria for FDG PET/CT in multiple myeloma: A new proposal from an Italian expert panel. IMPeTUs (Italian Myeloma criteria for PET USe). Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 414–421. [Google Scholar] [CrossRef]
- Flores-Montero, J.; de Tute, R.; Paiva, B.; Perez, J.J.; Böttcher, S.; Wind, H.; Sanoja, L.; Puig, N.; Lecrevisse, Q.; Vidriales, M.B.; et al. Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry Part B Clin. Cytom. 2016, 90, 61–72. [Google Scholar] [CrossRef]
- Stetler-Stevenson, M.; Paiva, B.; Stoolman, L.; Lin, P.; Jorgensen, J.L.; Orfao, A.; Van Dongen, J.; Rawstron, A.C. Consensus guidelines for myeloma minimal residual disease sample staining and data acquisition. Cytometry Part B Clin. Cytom. 2016, 90, 26–30. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; Van Der Velden, V.H.J.; Pérez-Morán, J.J.; Vidriales, M.B.; García-Sanz, R.; et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [Green Version]
- FDA. Authorizes First Next Generation Sequencing-Based Test to Detect Very Low Levels of Remaining Cancer Cells in Patients with Acute Lymphoblastic Leukemia or Multiple Myeloma. Available online: https://www.fda.gov/news-events/press-announcements/fda-authorizes-first-next-generation-sequencing-based-test-detect-very-low-levels-remaining-cancer (accessed on 23 October 2019).
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladetto, M.; Brüggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Bene, M.C.; Wuilleme, S.; Corre, J.; Attal, M.; Arnulf, B.; Garderet, L.; Macro, M.; Stoppa, A.-M.; Delforge, M.; et al. Concordance of Post-consolidation Minimal Residual Disease Rates by Multiparametric Flow Cytometry and Next-generation Sequencing in CASSIOPEIA. In Proceedings of the 17th International Myeloma Workshop, Boston, MA, USA, 12–15 September 2019; Volume 8. [Abstract #OAB–004]. [Google Scholar]
- Rawstron, A.C.; Gregory, W.M.; de Tute, R.M.; Davies, F.E.; Bell, S.E.; Drayson, M.T.; Cook, G.; Jackson, G.H.; Morgan, G.J.; Child, J.A.; et al. Minimal residual disease in myeloma by flow cytometry: Independent prediction of survival benefit per log reduction. Blood 2015, 125, 1932–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Cedena, M.T.; Puig, N.; Arana, P.; Vidriales, M.B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.L.; Martinez-Lopez, J.; et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Child, J.A.; de Tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal Residual Disease Assessed by Multiparameter Flow Cytometry in Multiple Myeloma: Impact on Outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef]
- Oliva, S.; op Bruinink, D.H.; ŘÍhová, L.; Spada, S.; van der Holt, B.; Troia, R.; Gambella, M.; Pantani, L.; Grammatico, S.; Gilestro, M.; et al. Minimal residual disease (MRD) monitoring by multiparameter flow cytometry (MFC) in newly diagnosed transplant eligible multiple myeloma (MM) patients: Results from the EMN02/HO95 phase 3 trial. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.T.; Cordon, L.; Vidriales, M.-B.; Burgos, L.; Flores-Montero, J.; Lopez-Anglada, L.; Gutierrez, N.; Calasanz, M.J.; et al. Impact of Next-Generation Flow (NGF) Minimal Residual Disease (MRD) Monitoring in Multiple Myeloma (MM): Results from the Pethema/GEM2012 Trial. Blood 2017, 130. Abstract #905 [ASH 2017 58th Meeting]. [Google Scholar] [CrossRef]
- Bladé, J.; Fernández De Larrea, C.; Rosiñol, L.; Cibeira, M.T.; Jiménez, R.; Powles, R. Soft-tissue plasmacytomas in multiple myeloma: Incidence, mechanisms of extramedullary spread, and treatment approach. J. Clin. Oncol. 2011, 29, 3805–3812. [Google Scholar] [CrossRef]
- Moreau, P. PET-CT in MM: A new definition of CR. Blood 2011, 118, 5984–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.-M.; et al. Prospective Evaluation of Magnetic Resonance Imaging and [18F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients With Multiple Myeloma Included in the IFM/DFCI 2009 Trial. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar] [CrossRef] [PubMed]
- Zamagni, E.; Patriarca, F.; Nanni, C.; Zannetti, B.; Englaro, E.; Pezzi, A.; Tacchetti, P.; Buttignol, S.; Perrone, G.; Brioli, A.; et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood 2011, 118, 5989–5995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, T.B.; Haessler, J.; Brown, T.L.Y.; Shaughnessy, J.D.; van Rhee, F.; Anaissie, E.; Alpe, T.; Angtuaco, E.; Walker, R.; Epstein, J.; et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood 2009, 114, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Zamagni, E.; Nanni, C.; Dozza, L.; Carlier, T.; Tacchetti, P.; Versari, A.; Chauvie, S.; Gallamini, A.; Attal, M.; Gamberi, B.; et al. Standardization of 18F-FDG PET/CT According to Deauville Criteria for MRD Evaluation in Newly Diagnosed Transplant Eligible Multiple Myeloma Patients: Joined Analysis of Two Prospective Randomized Phase III Trials. Blood 2018, 132. Abstract #257 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia 2019, 33, 1713–1722. [Google Scholar] [CrossRef]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.-L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; San-Miguel, J.F.; Casneuf, T.; Iida, S.; Lonial, S.; Usmani, S.Z.; Spencer, A.; Moreau, P.; Plesner, T.; Weisel, K.; et al. Evaluation of Sustained Minimal Residual Disease (MRD) Negativity in Relapsed/Refractory Multiple Myeloma (RRMM) Patients (Pts) Treated with Daratumumab in Combination with Lenalidomide Plus Dexamethasone (D-Rd) or Bortezomib Plus Dexamethasone (D-Vd): An. Blood 2018, 132. Abstract #3272 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Radich, J.P.; Deininger, M.; Abboud, C.N.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; Curtin, P.; DeAngelo, D.J.; Gotlib, J.; et al. Chronic myeloid leukemia, version 1.2019. JNCCN J. Natl. Compr. Cancer Netw. 2018, 16, 1108–1135. [Google Scholar] [CrossRef]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.J.W.M.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Liu, J.; Chen, M.; Huang, B.; Li, J. Longitudinal Flow Cytometry Identified “Minimal Residual Disease” (MRD) Evolution Patterns for Predicting the Prognosis of Patients with Transplant-Eligible Multiple Myeloma. Biol. Blood Marrow Transplant. 2018, 24, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Lakshman, A.; Vincent Rajkumar, S.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Cherry, B.M.; Korde, N.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Mulquin, M.; Zuchlinski, D.; Yancey, M.A.; Maric, I.; Calvo, K.R.; et al. Modeling progression risk for smoldering multiple myeloma: Results from a prospective clinical study. Proc. Leuk. Lymphoma 2013, 54, 2215–2218. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef]
- Pérez-Persona, E.; Vidriales, M.B.; Mateo, G.; García-Sanz, R.; Mateos, M.V.; De Coca, A.G.; Galende, J.; Martín-Nuñez, G.; Alonso, J.M.; De Heras, N.L.; et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007, 110, 2586–2592. [Google Scholar] [CrossRef]
- San Miguel, J.; Mateos, M.-V.; Gonzalez, V.; Dimopoulos, M.A.; Kastritis, E.; Hajek, R.; de Larrea Rodríguez, C.; Morgan, G.J.; Merlini, G.; Mangiacavalli, S.; et al. Updated risk stratification model for smoldering multiple myeloma (SMM) incorporating the revised IMWG diagnostic criteria. J. Clin. Oncol. 2019, 37. Abstract #8000 [ASCO 2019 Annual Meeting] Updated data presented at the meeting. [Google Scholar] [CrossRef]
- Gay, F.; Oliva, S.; Petrucci, M.T.; Conticello, C.; Catalano, L.; Corradini, P.; Siniscalchi, A.; Magarotto, V.; Pour, L.; Carella, A.; et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: A randomised, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 1617–1629. [Google Scholar] [CrossRef]
- López-Corral, L.; Gutiérrez, N.C.; Vidriales, M.B.; Mateos, M.V.; Rasillo, A.; García-Sanz, R.; Paiva, B.; San Miguel, J.F. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin. Cancer Res. 2011, 17, 1692–1700. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.V.; Hernández, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosiñol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Korde, N.; Roschewski, M.; Zingone, A.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; Kazandjian, D.; Mailankody, S.; Wu, P.; et al. Treatment With Carfilzomib-Lenalidomide-Dexamethasone With Lenalidomide Extension in Patients With Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol. 2015, 1, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateos, M.-V.; Martínez-López, J.; Rodríguez-Otero, P.; de la Calle, G.V.; González, M.-S.; Oriol, A.; Gutiérrez, N.-C.; Paiva, B.; Rios, R.; Rosiñol, L.; et al. Curative strategy (gem-cesar) for high-risk smoldering myeloma: Carfilzomib, lenalidomide and dexamethasone (krd) as induction followed by hdt-asct, consolidation with krd and maintenance with rd. HemaSphere 2019, 3, 390. [Google Scholar] [CrossRef]
- Mailankody, S.; Salcedo, M.; Tavitian, E.; Korde, N.; Lendvai, N.; Hassoun, H.; Lesokhin, A.M.; Lahoud, O.B.; Smith, E.L.; Hultcrantz, M.; et al. Ixazomib and dexamethasone in high risk smoldering multiple myeloma: A clinical and correlative pilot study. J. Clin. Oncol. 2019, 37. Abstract #8051 [ASCO 2019 Annual Meeting]. [Google Scholar] [CrossRef]
- Jagannath, S.; Laubach, J.; Wong, E.; Stockerl-Goldstein, K.; Rosenbaum, C.; Dhodapkar, M.; Jou, Y.-M.; Lynch, M.; Robbins, M.; Shelat, S.; et al. Elotuzumab monotherapy in patients with smouldering multiple myeloma: A phase 2 study. Br. J. Haematol. 2018, 182, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ghobrial, I.M.; Bustoros, M.; Reyes, K.; Hornburg, K.; Badros, A.Z.; Vredenburgh, J.J.; Boruchov, A.; Matous, J.V.; Caola, A.; et al. Phase II Trial of Combination of Elotuzumab, Lenalidomide, and Dexamethasone in High-Risk Smoldering Multiple Myeloma. Blood 2018, 132. Abstract #154 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Bustoros, M.; Liu, C.; Reyes, K.; Hornburg, K.; Guimond, K.; Styles, R.; Savell, A.; Berrios, B.; Warren, D.; Dumke, H.; et al. Phase II Trial of the Combination of Ixazomib, Lenalidomide, and Dexamethasone in High-Risk Smoldering Multiple Myeloma. Blood 2018, 132. Abstract #804 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Brighton, T.A.; Khot, A.; Harrison, S.J.; Ghez, D.; Weiss, B.M.; Kirsch, A.; Magen, H.; Gironella, M.; Oriol, A.; Streetly, M.; et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin. Cancer Res. 2019, 25, 3772–3775. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Cavo, M.; Chari, A.; Cohen, Y.C.; Spencer, A.; Voorhees, P.M.; Estell, J.; Sandhu, I.; Jenner, M.; Williams, C.; et al. Updated Results from the Phase 2 Centaurus Study of Daratumumab (DARA) Monotherapy in Patients with Intermediate-Risk or High-Risk Smoldering Multiple Myeloma (SMM). Blood 2018, 132. Abstract #1994 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Voorhees, P.M.; Goldschmidt, H.; Baker, R.I.; Bandekar, R.; Kuppens, S.; Neff, T.; Qi, M.; Dimopoulos, M.A. Randomized, open-label, phase 3 study of subcutaneous daratumumab (DARA SC) versus active monitoring in patients (Pts) with high-risk smoldering multiple myeloma (SMM): AQUILA. J. Clin. Oncol. 2018, 36. Abstract #TPS8062 (ASCO 2018 Annual Meeting]. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2019, 37. Abstract #8001 [ASCO 2019 Annual Meeting]. [Google Scholar] [CrossRef]
- Barlogie, B.; Jagannath, S.; Desikan, K.R.; Mattox, S.; Vesole, D.; Siegel, D.; Tricot, G.; Munshi, N.; Fassas, A.; Singhal, S.; et al. Total therapy with tandem transplants for newly diagnosed multiple myeloma. Blood 1999, 93, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Barlogie, B.; Mitchell, A.; van Rhee, F.; Epstein, J.; Yaccoby, S.; Zangari, M.; Heuck, C.; Hoering, A.; Morgan, G.J.; Crowley, J. Curing Multiple Myeloma (MM) with Total Therapy (TT). Blood 2014, 124. Abstract #195 [ASH 2014 56th Meeting]. [Google Scholar] [CrossRef]
- Mateos, M.V.; San Miguel, J.F. Management of multiple myeloma in the newly diagnosed patient. Hematology 2017, 2017, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Attal, M.; Facon, T. Frontline therapy of multiple myeloma. Blood 2015, 125, 3076–3084. [Google Scholar] [CrossRef]
- Harousseau, J.-L.; Attal, M.; Avet-Loiseau, H.; Marit, G.; Caillot, D.; Mohty, M.; Lenain, P.; Hulin, C.; Facon, T.; Casassus, P.; et al. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: Results of the IFM 2005-01 phase III trial. J. Clin. Oncol. 2010, 28, 4621–4629. [Google Scholar] [CrossRef]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wäsch, R.; Giaccone, L.; Auner, H.W.; Caers, J.; Gramatzki, M.; van de Donk, N.; Oliva, S.; et al. From transplant to novel cellular therapies in multiple myeloma: EMN guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef]
- Moreau, P.; Hulin, C.; Macro, M.; Caillot, D.; Chaleteix, C.; Roussel, M.; Garderet, L.; Royer, B.; Brechignac, S.; Tiab, M.; et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: Results of the prospective IFM2013-04 trial. Blood 2016, 127, 2569–2574. [Google Scholar] [CrossRef]
- Roussel, M.; Lauwers-Cances, V.; Robillard, N.; Hulin, C.; Leleu, X.; Benboubker, L.; Marit, G.; Moreau, P.; Pegourie, B.; Caillot, D.; et al. Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: A phase II study by the Intergroupe Francophone du Myélome. J. Clin. Oncol. 2014, 32, 2712–2717. [Google Scholar]
- Kumar, S.; Flinn, I.; Richardson, P.G.; Hari, P.; Callander, N.; Noga, S.J.; Stewart, A.K.; Turturro, F.; Rifkin, R.; Wolf, J.; et al. Randomized, multicenter, phase 2 study (EVOLUTION) of combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide in previously untreated multiple myeloma. Blood 2012, 119, 4375–4382. [Google Scholar] [CrossRef]
- Pawlyn, C.; Davies, F.; Cairns, D.; Striha, A.; Hockaday, A.; Kishore, B.; Garg, M.; Williams, C.; Karunanithi7, K.; Lindsay, J.; et al. Quadruplet KCRD (Carfilzomib, Cyclophosphamide, Lenalidomide and Dexamethasone) Induction for Newly Diagnosed Myeloma Patients. In Proceedings of the International Myeloma Workshop, Boston, MA, USA, 12–15 September 2019; p. e2, [Abstract #OAB–002]. Updated data presented at the meeting. [Google Scholar]
- Jackson, G.H.; Davies, F.E.; Pawlyn, C.; Cairns, D.; Striha, A.; Hockaday, A.; Collett, C.; Jones, J.R.; Kishore, B.; Garg, M.; et al. A Quadruplet Regimen Comprising Carfilzomib, Cyclophosphamide, Lenalidomide, Dexamethasone (KCRD) Vs an Immunomodulatory Agent Containing Triplet (CTD/CRD) Induction Therapy Prior to Autologous Stem Cell Transplant: Results of the Myeloma XI Study. Blood 2018, 132. Abstract #302 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Rosinol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, Lenalidomide and Dexamethasone (VRD-GEM) As Induction Therapy Prior Autologous Stem Cell Transplantation (ASCT) in Multiple Myeloma (MM): Results of a Prospective Phase III Pethema/GEM Trial. Blood 2017, 130. Abstract #2017 [ASH 2017 59th Annual Meeting]. [Google Scholar] [CrossRef]
- Voorhees, P.; Kaufman, J.L.; Laubach, J.; Sborov, D.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.; Anderson, L.; et al. Daratumumab + Lenalidomide, Bortezomib & Dexamethasone Improves Depth of Response in Transplant-eligible Newly Diagnosed Multiple Myeloma: GRIFFIN. In Proceedings of the 17th International Myeloma Workshop, Boston, MA, USA, 12–15 Septemebr 2019; pp. 546–547, [Abstract #OAB–87]. [Google Scholar]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Kumar, S.K.; Berdeja, J.G.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, A.K.; Hari, P.; Roy, V.; Vescio, R.; et al. Ixazomib, lenalidomide, and dexamethasone in patients with newly diagnosed multiple myeloma: Long-term follow-up including ixazomib maintenance. Leukemia 2019, 33, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Rosiñol Dachs, L.; Hebraud, B.; Oriol, A.; Colin, A.-L.; Rios, R.; Hulin, C.; Blanchard, M.J.; Caillot, D.; Sureda, A.; Hernández, M.T.; et al. Integrated Analysis of Randomized Controlled Trials Evaluating Bortezomib + Lenalidomide + Dexamethasone or Bortezomib + Thalidomide + Dexamethasone Induction in Transplant-Eligible Newly Diagnosed Multiple Myeloma. Blood 2018, 132. Abstract #3245 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Palumbo, A.; Cavallo, F.; Gay, F.; Di Raimondo, F.; Ben Yehuda, D.; Petrucci, M.T.; Pezzatti, S.; Caravita, T.; Cerrato, C.; Ribakovsky, E.; et al. Autologous Transplantation and Maintenance Therapy in Multiple Myeloma. N. Engl. J. Med. 2014, 371, 895–905. [Google Scholar] [CrossRef]
- Cavo, M.; Hájek, R.; Pantani, L.; Beksac, M.; Oliva, S.; Dozza, L.; Johnsen, H.E.; Petrucci, M.T.; Mellqvist, U.-H.; Conticello, C.; et al. Autologous Stem Cell Transplantation Versus Bortezomib-Melphalan-Prednisone for Newly Diagnosed Multiple Myeloma: Second Interim Analysis of the Phase 3 EMN02/HO95 Study. Blood 2017, 130. Abstract #397 [ASH 2017 59th Meeting]. [Google Scholar] [CrossRef]
- Cavo, M.; Goldschmidt, H.; Rosinol, L.; Pantani, L.; Zweegman, S.; Salwender, H.J.; Lahuerta, J.J.; Lokhorst, H.M.; Petrucci, M.T.; Blau, I.; et al. Double Vs Single Autologous Stem Cell Transplantation for Newly Diagnosed Multiple Myeloma: Long-Term Follow-up (10-Years) Analysis of Randomized Phase 3 Studies. Blood 2018, 132. Abstract #124 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Pasquini, M.C.; Blackwell, B.; Hari, P.; Bashey, A.; Devine, S.; Efebera, Y.; Ganguly, S.; Gasparetto, C.; Geller, N.; et al. Autologous transplantation, consolidation, and maintenance therapy in multiple myeloma: Results of the BMT CTN 0702 trial. J. Clin. Oncol. 2019, 37, 589–597. [Google Scholar] [CrossRef]
- Sonneveld, P.; Beksac, M.; Van der Holt, B.; Dimopoulos, M.; Carella, A.; Ludwig, H.; Driessen, C.; Wester, R.; Hajek, R.; Cornelisse, P.; et al. Consolidation followed by maintenance vs maintenance alone in newly diagnosed, transplant eligible multiple myeloma: A randomized phase 3 study of the european myeloma network (emn02/ho95 mm trial). HemaSphere 2018, 2, 5–6. [Google Scholar]
- McCarthy, P.L.; Holstein, S.A.; Petrucci, M.T.; Richardson, P.G.; Hulin, C.; Tosi, P.; Bringhen, S.; Musto, P.; Anderson, K.C.; Caillot, D.; et al. Lenalidomide Maintenance After Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: A Meta-Analysis. J. Clin. Oncol. 2017, 35, 3279–3289. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.H.; Davies, F.E.; Pawlyn, C.; Cairns, D.A.; Striha, A.; Collett, C.; Hockaday, A.; Jones, J.R.; Kishore, B.; Garg, M.; et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Oliva, S.; Gambella, M.; Larocca, A.; Spada, S.; Marzanati, E.; Mantoan, B.; Grammatico, S.; Conticello, C.; Gamberi, B.; Offidani, M.; et al. Prognostic Impact of Minimal Residual Disease By ASO-RQ-PCR in Multiple Myeloma: A Pooled Analysis of 2 Phase III Studies in Patients Treated with Lenalidomide after Front-Line Therapy. Blood 2016, 128. Abstract #4409 [ASH 2016 58th Meeting]. [Google Scholar] [CrossRef]
- Bonello, F.; Pulini, S.; Ballanti, S.; Gentile, M.; Spada, S.; Annibali, O.; Omedé, P.; Ronconi, S.; Cangialosi, C.; Podda, L.; et al. Lenalidomide Maintenance with or without Prednisone in Newly Diagnosed Myeloma Patients: A Pooled Analysis. Cancers 2019, 11, 1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mian, I.; Milton, D.R.; Shah, N.; Nieto, Y.; Popat, U.R.; Kebriaei, P.; Parmar, S.; Oran, B.; Shah, J.J.; Manasanch, E.E.; et al. Prolonged survival with a longer duration of maintenance lenalidomide after autologous hematopoietic stem cell transplantation for multiple myeloma. Cancer 2016, 122, 3831–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, P.; Avet-Loiseau, H.; Lonial, S.; Usmani, S.; Siegel, D.; Anderson, K.C.; Chng, W.-J.; Moreau, P.; Attal, M.; Kyle, R.A.; et al. Treatment of multiple myeloma with high-risk cytogenetics: A consensus of the International Myeloma Working Group. Blood 2016, 127, 2955–2962. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Lokhorst, H.M.; Mai, E.K.; van der Holt, B.; Blau, I.W.; Zweegman, S.; Weisel, K.C.; Vellenga, E.; Pfreundschuh, M.; Kersten, M.J.; et al. Bortezomib before and after high-dose therapy in myeloma: Long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 2018, 32, 383–390. [Google Scholar] [CrossRef]
- D’Agostino, M.; De Paoli, L.; Conticello, C.; Offidani, M.; Ria, R.; Petrucci, M.T.; Spada, S.; Marcatti, M.; Catalano, L.; Gilestro, M.; et al. Continuous therapy in standard- and high-risk newly-diagnosed multiple myeloma: A pooled analysis of 2 phase III trials. Crit. Rev. Oncol. Hematol. 2018, 132, 9–16. [Google Scholar] [CrossRef]
- Gay, F.; Jackson, G.; Rosiñol, L.; Holstein, S.A.; Moreau, P.; Spada, S.; Davies, F.; Lahuerta, J.J.; Leleu, X.; Bringhen, S.; et al. Maintenance Treatment and Survival in Patients With Myeloma. JAMA Oncol. 2018, 4, 1389. [Google Scholar] [CrossRef]
- D’Agostino, M.; Zaccaria, G.M.; Ziccheddu, B.; Genuardi, E.; Maura, F.; Oliva, S.; Auclair, D.; Yesil, J.; Capra, A.; Colucci, P.; et al. Clinical and Biological Early Relapse Predictors in Multiple Myeloma: An Analysis from the MMRF CoMMpass Study. In Proceedings of the 17 th International Myeloma Workshop, Boston, MA, USA, 12–15 September 2019; pp. 20–21, [Abstract #OAB–018]. [Google Scholar]
- Dimopoulos, M.A.; Gay, F.; Schjesvold, F.; Beksac, M.; Hajek, R.; Weisel, K.C.; Goldschmidt, H.; Maisnar, V.; Moreau, P.; Min, C.K.; et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2019, 393, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Jakubowiak, A.J.; Dytfeld, D.; Griffith, K.A.; Lebovic, D.; Vesole, D.H.; Jagannath, S.; Al-Zoubi, A.; Anderson, T.; Nordgren, B.; Detweiler-Short, K.; et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012, 120, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.; Raje, N.; Vij, R.; Reece, D.; Berdeja, J.; Vesole, D.; Jagannath, S.; Cole, C.; Faham, M.; Nam, J.; et al. Improved Efficacy After Incorporating Autologous Stem Cell Transplant (Asct) Into Krd Treatment With Carfilzomib (Cfz), Lenalidomide (Len), And Dexamethasone (Dex) In Newly Diagnosed Multiple Myeloma. Haematologica 2016, 101, 1–2. [Google Scholar]
- Roussel, M.; Lauwers-Cances, V.; Robillard, N.; Belhadj, K.; Facon, T.; Garderet, L.; Escoffre, M.; Pegourie, B.; Benboubker, L.; Caillot, D.; et al. Frontline Therapy with Carfilzomib, Lenalidomide, and Dexamethasone (KRd) Induction Followed By Autologous Stem Cell Transplantation, Krd Consolidation and Lenalidomide Maintenance in Newly Diagnosed Multiple Myeloma (NDMM) Patients: Primary Results of th. Blood 2016, 128. Abstract #1142 [ASH 2017 59th Meeting]. [Google Scholar] [CrossRef]
- Gay, F.; Cerrato, C.; Scalabrini, D.R.; Galli, M.; Belotti, A.; Zamagni, E.; Ledda, A.; Grasso, M.; Angelucci, E.; Liberati, A.M.; et al. Carfilzomib-Lenalidomide-Dexamethasone (KRd) Induction-Autologous Transplant (ASCT)-Krd Consolidation Vs KRd 12 Cycles Vs Carfilzomib-Cyclophosphamide-Dexamethasone (KCd) Induction-ASCT-KCd Consolidation: Analysis of the Randomized FORTE Trial in Newly Di. Blood 2018, 132. Abstract #121 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Weisel, K.; Asemissen, A.M.; Schieferdecker, A.; Besemer, B.; Zago, M.; Mann, C.; Lutz, R.; Benner, A.; Tichy, D.; Bokemeyer, C.; et al. Isatuximab, Carfilzomib, Lenalidomide and Dexamethasone (I-KRd) in front-line treatment of high-risk Multiple Myeloma: Results of the Safety Run-In cohort in the phase II, multicenter GMMG-CONCEPT trial. In Proceedings of the International Myeloma Workshop, Boston, MA, USA, 12–15 September 2019; pp. 25–26, [Abstract #OAB–023]. [Google Scholar]
- Bringhen, S.; D’Agostino, M.; Paris, L.; Ballanti, S.; Pescosta, N.; Spada, S.; Pezzatti, S.; Grasso, M.; Rota-Scalabrini, D.; De Rosa, L.; et al. Lenalidomide-based induction and maintenance in elderly newly diagnosed multiple myeloma patients: Updated results of the EMN01 randomized trial. Haematologica 2019. [ahead of print]. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvini, M.; D’Agostino, M.; Bonello, F.; Boccadoro, M.; Bringhen, S. Determining treatment intensity in elderly patients with multiple myeloma. Expert Rev. Anticancer Ther. 2018, 18, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Bringhen, S.; Mateos, M.-V.; Larocca, A.; Facon, T.; Kumar, S.; Offidani, M.; McCarthy, P.; Evangelista, A.; Lonial, S.; et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: An International Myeloma Working Group report. Blood 2015, 125, 2068–2074. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus Melphalan and Prednisone for Initial Treatment of Multiple Myeloma. N. Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [Green Version]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Persistent Overall Survival Benefit and No Increased Risk of Second Malignancies With Bortezomib-Melphalan-Prednisone Versus Melphalan-Prednisone in Patients With Previously Untreated Multiple Myeloma. J. Clin. Oncol. 2013, 31, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Dimopoulos, M.A.; Dispenzieri, A.; Catalano, J.V.; Belch, A.; Cavo, M.; Pinto, A.; Weisel, K.; Ludwig, H.; Bahlis, N.J.; et al. Final analysis of survival outcomes in the phase 3 FIRST trial of up-front treatment for multiple myeloma. Blood 2018, 131, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Larocca, A.; Salvini, M.; De Paoli, L.; Cascavilla, N.; Benevolo, G.; Galli, M.; Montefusco, V.; di Toritto, T.C.; Baraldi, A.; Spada, S.; et al. Efficacy and Feasibility of Dose/Schedule-Adjusted Rd-R Vs. Continuous Rd in Elderly and Intermediate-Fit Newly Diagnosed Multiple Myeloma (NDMM) Patients: RV-MM-PI-0752 Phase III Randomized Study. Blood 2018, 132. Abstract #305 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Durie, B.G.M.; Hoering, A.; Abidi, M.H.; Rajkumar, S.V.; Epstein, J.; Kahanic, S.P.; Thakuri, M.; Reu, F.; Reynolds, C.M.; Sexton, R.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): A randomised, open-label, phase 3 trial. Lancet 2017, 389, 519–527. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, E.K.; Laubach, J.P.; Yee, A.J.; Chen, T.; Huff, C.A.; Basile, F.G.; Wade, P.M.; Paba-Prada, C.E.; Ghobrial, I.M.; Schlossman, R.L.; et al. A phase 2 study of modified lenalidomide, bortezomib and dexamethasone in transplant-ineligible multiple myeloma. Br. J. Haematol. 2018, 182, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Otero, P.R.; Bringhen, S.; Oliva, S.; Nogai, A.; Attal, M.; Moreau, P.; Kanagavel, D.; Fitzmaurice, T.; Wu, J.; et al. Preliminary Results from a Phase I Study of Isatuximab (ISA) in Combination with Bortezomib, Lenalidomide, Dexamethasone (VRd) in Patients with Newly Diagnosed Multiple Myeloma (NDMM) Non-Eligible for Transplant. Blood 2018, 132. Abstract #595 [ASH 2018 60th Meeting]. [Google Scholar] [CrossRef]
- Moreau, P.; Kolb, B.; Attal, M.; Caillot, D.; Benboubker, L.; Tiab, M.; Touzeau, C.; Leleu, X.; Roussel, M.; Chaleteix, C.; et al. Phase 1/2 study of carfilzomib plus melphalan and prednisone in patients aged over 65 years with newly diagnosed multiple myeloma. Blood 2015, 125, 3100–3104. [Google Scholar] [CrossRef] [Green Version]
- Facon, T.; Lee, J.H.; Moreau, P.; Niesvizky, R.; Dimopoulos, M.; Hajek, R.; Pour, L.; Jurczyszyn, A.; Qiu, L.; Klippel, Z.; et al. Carfilzomib or bortezomib with melphalan-prednisone for transplant-ineligible patients with newly diagnosed multiple myeloma. Blood 2019, 133, 1953–1963. [Google Scholar] [CrossRef]
- Bringhen, S.; Petrucci, M.T.; Larocca, A.; Conticello, C.; Rossi, D.; Magarotto, V.; Musto, P.; Boccadifuoco, L.; Offidani, M.; Omede, P.; et al. Carfilzomib, cyclophosphamide, and dexamethasone in patients with newly diagnosed multiple myeloma: A multicenter, phase 2 study. Blood 2014, 124, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Bringhen, S.; D’Agostino, M.; De Paoli, L.; Montefusco, V.; Liberati, A.M.; Galieni, P.; Grammatico, S.; Muccio, V.E.; Esma, F.; De Angelis, C.; et al. Phase 1/2 study of weekly carfilzomib, cyclophosphamide, dexamethasone in newly diagnosed transplant-ineligible myeloma. Leukemia 2018, 32, 979–985. [Google Scholar] [CrossRef]
- Bringhen, S.; Mina, R.; Petrucci, M.T.; Gaidano, G.; Ballanti, S.; Musto, P.; Offidani, M.; Spada, S.; Benevolo, G.; Ponticelli, E.; et al. Once-weekly versus twice-weekly carfilzomib in patients with newly diagnosed multiple myeloma: A pooled analysis of two phase I/II studies. Haematologica 2019, 104, 1640–1647. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Heuck, C.; Mitchell, A.; Szymonifka, J.; Nair, B.; Hoering, A.; Alsayed, Y.; Waheed, S.; Haider, S.; Restrepo, A.; et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012, 97, 1761–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granell, M.; Calvo, X.; Garcia-Guiñón, A.; Escoda, L.; Abella, E.; Martínez, C.M.; Teixidó, M.; Gimenez, M.T.; Senín, A.; Sanz, P.; et al. Prognostic impact of circulating plasma cells in patients with multiple myeloma: Implications for plasma cell leukemia definition. Haematologica 2017, 102, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneveld, P. Management of multiple myeloma in the relapsed/refractory patient. Hematology 2017, 2017, 508–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Rustad, E.H. Meeting report: Advances in minimal residual disease testing in multiple myeloma 2018. Adv. Cell Gene Ther. 2019, 2, e26. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, H.; Takezako, N.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Kong, K.A.; Murata, R.; Ito, S.; Miyamoto, T.; Yokoyama, K.; et al. Prognostic value of sequencing-based minimal residual disease detection in patients with multiple myeloma who underwent autologous stem-cell transplantation. Ann. Oncol. 2017, 28, 2503–2510. [Google Scholar] [CrossRef]
- Hahn, T.E.; Wallace, P.K.; Fraser, R.; Fei, M.; Tario, J.D.; Howard, A.; Zhang, Y.; Blackwell, B.; Brunstein, C.G.; Efebera, Y.A.; et al. Minimal Residual Disease (MRD) Assessment before and after Autologous Hematopoietic Cell Transplantation (AutoHCT) and Maintenance for Multiple Myeloma (MM): Results of the Prognostic Immunophenotyping for Myeloma Response (PRIMeR) Study. Biol. Blood Marrow Transplant. 2019, 25, S4–S6. [Google Scholar] [CrossRef]
- Paiva, B.; Vídriales, M.B.; Rosiñol, L.; Martínez-López, J.; Mateos, M.V.; Ocio, E.M.; Montalbán, M.A.; Cordón, L.; Gutiérrez, N.C.; Corchete, L.; et al. A multiparameter flow cytometry immunophenotypic algorithm for the identification of newly diagnosed symptomatic myeloma with an MGUS-like signature and long-term disease control. Leukemia 2013, 27, 2056–2061. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Characteristics | NGS | NGF |
|---|---|---|
| Applicability | ≥90% | ~100% |
| Baseline sample | Required for molecular marker identification | Not required |
| Number of cells | 1–2 million cells/20 µg DNA | 10 million cells/tube |
| Standardization | Commercial companies (e.g., Adaptive Biotechnologies) | Euro Flow consortium |
| Sample processing | Fresh and/or stored samples | Fresh sample required; processing within ≤48 h |
| Sample quality control | Not feasible | Possible to check by global bone marrow cell analysis |
| Quantitative | Yes | Yes |
| Sensitivity | 1 in 10−5–10−6 | 1 in 10−5–10−6 |
| Clonal evolution | Evaluable | Not evaluable |
| Time required | 1 week | 3–4 h |
| Support required | Bioinformatics support | Expert flow cytometrist; Automated software |
| Stratification System | N | Low Risk | Intermediate Risk | High Risk | |||||
|---|---|---|---|---|---|---|---|---|---|
| Criteria | TTP (Median) | Criteria | TTP (Median) | Criteria | TTP (Median) | ||||
| Mayo Clinic [47] | 273 | - M-protein <3 g/dL plus - BMPCs <10% | 19 years | - M-protein <3 g/dL plus - BMPCs ≥10% | 8 years | - M-protein ≥3 g/dL plus - BMPCs ≥10% | 2 years | ||
| PETHEMA [48] | 89 | - Aberrant PCs by MFC <95% - Immunoparesis | NR | - Aberrant PCs by MFC ≥95% OR - Immunoparesis | 6 years | - Aberrant PCs by MFC ≥95% AND - Immunoparesis | 1.9 years | ||
| Mayo 2/20/20 [45] | 421 | None of the risk factors: - M-protein >2g/dL - BMPCs >20% - FLCr >20 | 9.1 years | One risk factor: - M-protein >2g/dL - BMPCs >20% - FLCr >20 | 5.6 years | ≥2 risk factors: - M-protein >2g/dL - BMPCs >20% - FLCr >20 | 2.4 years | ||
| Low Risk | Intermediate Low | Intermediate High | High Risk | ||||||
| N | Criteria | 2-year progression risk | Criteria | 2-year progression risk | Criteria | 2-year progression risk | Criteria | 2-year progression risk | |
| 2/20/20 + CA by FISH * [49] | 2004 | None of the risk factors: - M-protein >2g/dL - FLCr >20 - BMPCs >20% Presence of any of the CA * | 8% | 1 risk factor: - M-protein >2g/dL - FLCr >20 - BMPCs >20% Presence of any of the CA * | 21% | 2 risk factors: - M-protein >2g/dL - FLCr >20 - BMPCs >20% Presence of any of the CA * | 37% | ≥3 risk factors: - M-protein >2g/dL - FLCr >20 - BMPCs >20% Presence of any of the CA * | 59% |
| Protocol | Phase | Treatment | Pts | Response | TTP/PFS/OS | Toxicity (≥G3) | |
|---|---|---|---|---|---|---|---|
| NCT00480363 QuiRedex [52] | Phase III | Rd vs. observation | Induction: 28-day cycle (cycles 1–9) lenalidomide 25 mg p.o. days (D) 1–21 + dex 20 mg p.o. D1–4, 12–15 Maintenance: (cycles 1–24) 28-day cycle Lenalidomide 10 mg D1–21 vs. Observation | 119 | FU 73 months Best response 26% CR after maintenance | Median TTP NR (HR 0.24) 3-year PFS 77% 3-, 5-year OS 94%, 88% (HR 0.43) vs. median TTP 23 months (p < 0.001) 3-year PFS 30% (p < 0.001) 3-, 5-year OS 80%, 71%, (p = 0.03) | Neutropenia 5% Thrombocytopenia 2% Anemia 2% Infection 6% Asthenia 6% Skin rash 3% vs. none |
| NCT01572480 120107 [53] | Phase II | KRd induction Rd maintenance | Induction: 28-day cycle (cycles 1–8) carfilzomib 20/36 mg/m2 iv D1, 2, 8, 9, 15, 16 + lenalidomide 25 mg p.o. D 1–21 + dex 20 mg (cycles 1–4) and 10 (cycles 5–8) p.o. or iv D1, 2, 8, 9, 15, 16 Maintenance (cycles 1–24): 28-day cycle lenalidomide 25 mg D1–21 | 12 | CR 100%. MRD neg (MFC 10−5 ) 92% | No progression to MM 3-, 4-year PFS 94%, 70.6% 3-, 4- year OS: 100% | Hematologic: Lymphopenia 39% Neutropenia 28% Anemia 22% Thrombocytopenia 11% Non-hematologic: Diarrhea 17% Lung infection 17% Hypophosphatemia 11% 1 case of CHF |
| NCT02415413 GEM-CESAR [54] | Phase II | KRd induction ASCT KRd consolidation Rd maintenance | Induction: 28-day cycle (cycles 1–6): carfilzomib 20/36 mg/m2 iv D1, 2, 8, 9, 15, 16 + lenalidomide 25 mg p.o. D1–21 + dex 40 mg D1, 8, 15, 22 ASCT melphalan 200 mg/m2 Consolidation: KRd 2 cycles Maintenance: 28-day cycle lenalidomide 10 mg days 1–21 + 20 mg days 1, 8, 15, 22 for 2 years | 90 | ORR: 100%. ≥CR: 70% MRD (NGF) 57% | 30 months PFS 98% | Induction (G3-4): Neutropenia 6% Thrombocytopenia 11% Infections 18% Consolidation: Ongoing |
| NCT03673826 HO147SMM | Phase II | KRd vs. Rd Rd maintenance | Induction: (cycles 1–9) carfilzomib 20/36 mg/m2 iv D1, 2, 8, 9, 15, 16, lenalidomide 25 mg p.o. D1–21, dexamethasone 20 mg p.o. (cycles 1–4) and 10 mg p.o. (cycles 5–9) D1, 8, 15, 22 vs. (cycles 1–9) lenalidomide 25 mg p.o D1–21, dexamethasone 20 mg p.o. (cycles 1–4) and 10 mg p.o. (cycles 5–9) D1, 8, 15, 22 Maintenance: (cycles 1–24) lenalidomide 10 mg p.o. days 1–21 of a 28-day cycle | 120 | NA | NA | NA |
| NCT03289299 ASCENT | Phase II | Dara KRd + Rd maintenance | Induction: 28-day cycle (cycles 1–6) carfilzomib 20/36 mg/m2 iv D1, 2, 8, 9, 15, 16 + lenalidomide 25 mg p.o. D1–21 + dex 40 mg D1, 8, 15, 22, daratumumab 16 mg/kg iv D1, 8, 15, and 22 of cycles 1–2; D1 and 15 of cycles 3–6; Consolidation: 6 cycles, KR as in induction + dex 20 mg p.o. D1, 8, 15, 22 Daratumumab 16 mg/kg iv D1 of cycles 7–12 vs. ASCT Maintenance: 12 cycles, lenalidomide 10 mg p.o. D1–21 + daratumumab; 16 mg/kg iv D1 of odd cycles for cycles 13–24 | 83 | NA | NA | NA |
| NCT02697383 15-294 [55] | Phase I | Ixazomib dexamethasone | 28-day cycle (cycles 1–12) ixazomib 4 mg on D1, 8, and 15, and dexamethasone on days 1, 8, 15, and 22 (40 mg/week the first 4 cycles, thereafter 20 mg/week) | 14 | ORR 64% | 64% ORR (8 PR, and 1 VGPR) no patient progressed to MM | Lung infection (14%) |
| NCT01441973 CA204-011 [56] | Phase II | Elotuzumab | (cycle 1) elotuzumab 20 mg/kg iv D1, 8 then [cycle 2—progressive disease] Elotuzumab monthly q4 week vs. (cycle 1) elotuzumab 10 mg/kg iv D1, 8, 15, 22 (cycle 2—progressive disease) elotuzumab monthly q2 week | 31 | Both groups ORR 10% | Both groups 2-year PFS 69% | Upper respiratory tract infection 7% vs. Fatigue 6% Diarrhea 6% Insomnia 6% |
| NCT02279394 14-338 [57] | Phase II | Elotuzumab Rd + stem-cell mobilization + maintenance | Induction: 28-day cycle (cycles 1–2) elotuzumab 10 mg/kg iv D1, 8, 15, 22 + lenalidomide 25 mg p.o. D1–21 + dex 40 mg p.o. D1, 8, 15, 22 (cycles 3–8): elotuzumab 10 mg/kg iv D1, 15 + lenalidomide as in cycles 1–2 + dex 40 mg p.o. D1, 8, 15 Maintenance: 28-day cycle (cycles 9–4) elotuzumab 10 mg/kg iv D 1 + lenalidomide as in cycles 1–2 | 50 | ≥VGPR 43% | NA | Hypophosphatemia 34% Neutropenia 26% Lymphopenia 22% |
| NCT02916771 16-313 [58] | Phase II | Ixazomib-Rd | Induction: 28-day cycle (cycles 1–9): ixazomib 4 mg p.o. D1, 8, 15 + lenalidomide 25 mg p.o. D1–21 + dex 40 mg p.o. D1, 8, 15, 22 Maintenance: 28-day cycle (cycles 10-24): ixazomib 4 mg p.o. D1, 8, 15 + lenalidomide 25 mg p.o. D1–21 | 26 | ≥VGPR 53.8% | NA | Hypophosphatemia 13% Lymphopenia 13% Neutropenia 8.7% (G4 Neutropenia in 1 pt) |
| NCT01484275 CR100755 [59] | Phase II | Siltuxumab vs. placebo | (cycle 1—until progressive disease): siltuximab 15 mg/kg iv every 4 weeks vs. observation | 87 | NA | 1-year PFS: 84% Median PFS: NR vs. 1-year PFS: 74.4% Median PFS: 23.8 months | Infections (5 patients in siltuximab group and 6 patients in placebo group) Urinary disorders (one patient in the siltuximab group and three patients in the placebo group) |
| NCT02960555 2015-0148 | Phase II | Isatuximab | (cycles 1–30): 28-day cycle isatuximab iv D1, 8, 15, and 22 of cycle 1, on D1 and 15 of cycles 2–6, and on D1 of subsequent courses | 61 | NA | NA | NA |
| NCT02316106, CENTAURUS [60] | Phase II | Daratumumab iv (3 arms, 41 patients each) | Daratumumab 16 mg/kg iv in 8-week cycles Long intensity:(cycle 1) every 1 week; (cycle 2–3) every other week; (cycles 4–7) every 4 weeks; (cycles 8–20) every 8 weeks Intermediate intensity: (cycle 1) every 1 week and (cycles 2–20) every 8 weeks Short intensity: (cycle 1) every week for 8 infusions | 123 | ≥CR rate 7% in combined Long and Int arms | Long intensity 24-month PFS 90% vs. intermediate intensity 24-month PFS 82% vs. short intensity 24-month PFS 75% | Long intensity serious adverse events, 32% ≥G3 AE 44% - Hypertension 7% - Hyperglycemia 2% vs. intermediate intensity serious adverse events 15% ≥G3 AE 27% - Hypertension 5% - Hyperglycemia 5% vs. short intensity serious adverse events, 10% ≥G3 AE 15% - Hypertension 3% - Hyperglycemia 0% |
| NCT03301220 AQUILA [61] | Phase III | Daratumumab sc for 3 years vs. observation | Daratumumab sc injection (daratumumab 1800 mg + rHuPH20 [2000 U/mL]) once weekly for cycles 1 and 2 (D1, 8, 15, and 22 of each week), every 2 weeks for cycles 3–6 (D1 and 15), and thereafter every 4 weeks (D1) until 39 cycles or up to 36 months or PD vs. observation | 390 | NA | NA | NA |
| NCT01169337 E3A06 [62] | Phase II–III | R vs. observation (median FU 35 months) | Lenalidomide 25 mg p.o. D1-21 in 28 days cycle until PD vs. observation | 180 | Overall response 47.7% phase II 48.9% phase III | PFS 1 year (98% vs. 89%), PFS 2 years (93% vs. 76%) PFS 3 years (91% vs. 66%) | G3/4 non-hematologic AEs 28%: - Infections 10% - Fatigue 6.8% - Hypertension 9% G4 hematologic AEs 4.5%, primarily neutropenia (n = 4), cumulative incidence of invasive SPMs 11.4% (Len) and 3.5% (observation). |
| Protocol | Phase | Treatment | Subjects | Response | TTP/PFS/OS | Toxicity (≥G3) | |
|---|---|---|---|---|---|---|---|
| NCT01206205 IFM2008 [70] | Phase II | VRd induction Stem-cell mobilization ASCT VRd consolidation R maintenance | Induction 21-day cycles (cycles 1–3) Bortezomib iv 1.3 mg/m2 D1, 4, 8, and 11 Lenalidomide p.o. 25 mg D1–14 Dexamethasone p.o. 40 mg D1, 8, 15 Stem-cell harvest Consolidation 21-day cycles (cycles 1–3) Bortezomib iv 1.3 mg/m2 D1, 4, 8, and 11 Lenalidomide p.o. 25 mg D1–14 + dex p.o. 40 mg D1, 8, 15 Maintenance Lenalidomide p.o. 10 mg per day for the first 3 months, a possible dose increase to 15 mg for 1 year | 31 | After induction 58% ≥VGPR 23 % ≥CR 16% MRD neg MFC (10−4–10−5) After ASCT 70% ≥VGPR 47%≥CR 54% MRD neg MFC (10−4–10−5) After consolidation 87% ≥VGPR 50 ≥CR 58% MRD neg MFC (10−4–10−5) Best response (after maintenance) 84% ≥VGPR 58% ≥ CR 68% MRD neg MFC (10−4–10−5) | Estimated 3-year PFS 77% and OS 100% (median FU 39 months) | Neutropenia 65% Thrombocytopenia 19% Anemia 3% Infections 6% |
| NCT00507442 EVOLUTION [71] | Phase II randomized | VRd (n = 42) vs. VRCd (n = 48) vs. VCd (n = 33) | Induction 21-day cycles (cycles 1–8) Bortezomib 1.3 mg/m2, days 1, 4, 8, 11 Dexamethasone 40 mg, D1, 8, 15 Cyclophosphamide 500 mg/m2, D1, 8 Lenalidomide p.o., 15 mg in VRCd and 25 mg in VRd D1–14 Maintenance 6-week cycles (cycles 1–4) Bortezomib 1.3 mg/m2, D1, 8, 15, 22 Stem-cell mobilization any time after 2 cycles ASCT any time after 4 cycles | 140 | VRd best response 61% ≥VGPR 21% CR VRCd best response 54% ≥VGPR 21% CR VCd best response 24% ≥VGPR 10% CR | 1-year PFS 83% VRd 86% VRCd 93% VCd 100% VCd-mod 1-year OS estimate 92% VRCd arm 100% for the other three arms | VRd Neuropathy 17% vs. VRCd Neuropathy 13% vs. VCd Neuropathy 9% |
| NCT01554852 MyelomaXI [72,73] | Phase III randomized | Transplant eligible: CTd vs. CRd vs. KCRd induction CVd intensification vs. no intensification Mel200 ASCT Lenalidomide maintenance vs. observation | Transplant eligible: Induction CTd: 21-day cycle cyclophosphamide 500 mg p.o. D1, 8, 15 thalidomide 50 mg p.o. continuously D1–21 dexamethasone 40 mg p.o. D1–4, 12–15 CRd: 28-day cycle cyclophosphamide 500 mg p.o. D1, 8 lenalidomide 25 mg p.o. D1–21 dexamethasone 40 mg p.o. D1–4, 12–15 KCRd: 28-day cycle carfilzomib 36 mg/m2 i.v. D1, 2, 8, 9, 15, and 16 cyclophosphamide 500 mg p.o. D1, 8 lenalidomide 25 mg p.o. D1–21 dexamethasone 40 mg p.o. D1–4, 8, 9, 15, and 16 Consolidation CVd: 21-day cycles Bortezomib i.v. 1.3 mg/m2 D1, 4, 8, and 11 cyclophosphamide 500 mg p.o. D1, 8, and 15 Dexamethasone p.o. 20 mg D1, 2, 4, 5, 8, 9, 11, and 12 Maintenance Lenalidomide p.o. 10 mg D1–21 | 1056 | Post-induction CTd 53% ≥VGPR 11% MRD neg (MFC 10−4–10−5) CRd 65% ≥VGPR 21% MRD neg KCRd 82% ≥VGPR 55% MRD neg | 3-year PFS 50.3% CTd/CRd 64.5% KCRd | - Neutropenia 12.8% CTd 22.3% CRd 16.4% KCRd - Thrombocytopenia 1.2% CTd 2.3% CRd 8.4% KCRd |
| NCT01191060 IFM2009 [74] | Phase III | VRd induction arm VRd consolidation arm ASCT | Induction: 21-day cycles (cycles 1–3) Bortezomib i.v. 1.3 mg/m2 D1, 4, 8, and 11 Lenalidomide p.o. 25 mg D1–14 Dexamethasone p.o. 20 mg D1, 2, 4, 5, 8, 9, 11, and 12 Stem-cell mobilization with cyclophosphamide and G-CSF Consolidation (cycles 4–8) VRd reduced dex 10 mg (VRd-alone group) vs. Melphalan 200 mg/m2 ASCT + 2 cycles of VRd reduced dex 10 mg (transplantation group) Maintenance Lenalidomide p.o. 10 mg per day for the first 3 months, a possible dose increase to 15 mg for 1 year | 700 | VRd arm After induction 45% ≥VGPR After consolidation ≥69% VGPR Best response (after maintenance) 77% ≥VGPR 48% CR of which 65% MRD neg vs. VRd–ASCT After induction 47% ≥VGPR After consolidation 78% ≥VGPR Best response 88% ≥VGPR 59% CR of which 79% MRD neg | Median PFS 50 vs. 36 months OS 4years 81% vs. 82% | Neutropenia (47% vs. 92%) Gastrointestinal disorders (7% vs. 28%) Infections (9% vs. 20%) |
| NCT01916252 GEM2012MENOS65 [75] | Phase III | VRd 6 cycles Stem cell mobilization after 3 induction cycles Mel200-ASCT vs. BuMel-ASCT VRd 2 cycles post-transplant consolidation | Induction (cycles 1–6) Bortezomib 1.3 mg/m2 sc D1, 4, 8, 11 Lenalidomide 25 mg/day D1–21 Dexamethasone 40 mg on D1–4 and 9–12 at 4 weeks ASCT Melphalan 200 mg/m2 iv D 2 vs. Busulfan 9.6 mg/kg + Melphalan 140 mg/m2 Consolidation (cycles 7–8) same schedule as induction | 458 | After induction 29% VGPR 39% CR 28% sCR 34% MRD-neg (by NGF) After ASCT 27% VGPR 49% CR 36% sCR 53% MRD neg After consolidation 58% MRD neg | NA | Induction Neutropenia 11% Thrombocytopenia 6% Hepatic 4% Skin 3% Neuropathy 1% |
| NCT01029054 UMCC 2009.056 [8] | Phase I/II Phase II extended | Phase I KRd without ASCT induction and KRd maintenance Phase II KRd induction Mobilization ASCT/Mel200 KRd consolidation KRd maintenance | Phase I/II Induction (cycles 1–8) Carfilzomib iv 20/36 mg/m2 D1, 2, 8, 9, 15, 16 (20 mg/m2 given on cycle 1, D1) Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg/week Maintenance (cycles 9–24) Carfilzomib 36 mg/m2 D1, 2, 15, 16 Lenalidomide p.o. same dose of C8 D1–21 Dexamethasone p.o. 20 mg weekly Phase II Induction (cycles 1–4) Carfilzomib iv 20/36 mg/m2 D1, 2, 8, 9, 15, 16 (20 mg/m2 given on cycle 1, D1) Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg/week Stem-cell collection using G-CSF and plerixafor ASCT Melphalan 200 mg/m2 conditioning Consolidation (cycles 5–8) Carfilzomib i.v. 36 mg/m2 D1, 2, 8, 9, 15, 16 Lenalidomide p.o. 15 mg D1–21 (with option to escalate to 25 mg) Dexamethasone p.o. 20 mg weekly Maintenance (cycles 9–18) Carfilzomib 36 mg/m2 D1, 2, 15, 16 Lenalidomide p.o. same dose of cycle 8 D1–21 Dexamethasone p.o. 20 mg weekly Lenalidomide as single-agent off-study after C18. | 53 (phase I/II) 76 (phase II) | Phase I/II Post-induction (C8) 55% sCR Phase II Post-consolidation (C8) 96% ≥VGPR, 73% CR 69% sCR. Post-consolidation (C8) 82% MRD neg (MFC) (N = 33) 66% MRD neg (NGS) (N = 29) Post-Maintenance (C18) 90% MRD neg (MFC) (N = 20) 71% MRD neg (NGS) (N = 16) | Phase I/II 4- year PFS 63% 4-year OS 93% Phase II 2-year PFS 97% and 2-year OS 99% (median FU 17 months) | Phase II Lymphopenia 28% Neutropenia 18% Infections 8% |
| NCT01402284 110221 [53] | Phase II | KRd induction R maintenance | Induction (cycles 1–8) Carfilzomib iv 20/36 mg/m2, D1, 2, 8, 9, 15, 16 Lenalidomide 25 mg p.o. D1–21 Dexamethasone 20 mg (cycles 1–4) and 10 (cycles 5–8) p.o. or iv D1, 2, 8, 9, 15, 16 then Maintenance (cycles 1–24) lenalidomide 25 mg D1–21 | 45 | 62% MRD neg (calculated on NGS-evaluable NDMM patients) | 18-month PFS: 100% vs. 84% | Lymphopenia 34% Thrombocytopenia 11% Neutropenia 15% Anemia 12% Infection 6% Cardiac 5% Vascular 6% |
| NCT02203643 FORTE [9] | Phase II | KCd-ASCT-KCd (arm A), KRd-ASCT- KRd (arm B), KRd 12 cycles (arm C) | Induction (cycles 1–4) Carfilzomib 36 mg/m2 D1, 2, 8, 9, 15, and 16 of a 28-day cycle; lenalidomide p.o. 25 mg D1–21 Dexamethasone 20 mg D1, 2, 8, 9, 15, and 16) or Cyclophosphamide 300 mg/m2 on D1, 8, and 15 Intensification ASCT/mel200 or 4 KRd cyles (cycles 4–8) Consolidation (cycles 8–12) 4 cycles same as induction Maintenance KR vs. R (random) | 474 | Pre-maintenance response rates: ≥VGPR: 76% Arm A 89% Arm B 87% Arm C sCR: 32% Arm A 44 % Arm B 43 % Arm C Pre-maintenance MRD neg MFC 42% Arm A 58% Arm B 54% Arm C | NA | Dermatologic 1–13% Neutropenia 15–20% Thrombocytopenia 8–15% Infections 12–14% Increased liver enzymes 1–10% Hypertension 3–8% Cardiac 2–3% |
| NCT02874742 GRIFFIN [76] | Phase II | Dara-VRd induction Mobilization ASCT Dara-VRd consolidation Dara-R maintenance | Induction (cycles 1–6) Lenalidomide 25 mg p.o. D1–14 Bortezomib 1.3 mg/m2 sc D1, 4, 8, and 11 Dexamethasone 40 mg weekly Daratumumab 16 mg/kg iv D1, 8, and 15 of cycles 1–4 and on D1 of cycles 5–6. Maintenance (C7–32) lenalidomide 10 mg p.o. daily (15 mg beginning at cycle 10 if tolerated) on D1–21 every 28 days and Dara 16 mg/kg iv every 56 days; Maintenance lenalidomide may be continued beyond C32 | 207 (safety run in 16 pts) | Post-consolidation (Dara–VRd) 91% ≥VGPR 52% ≥CR of which 59% MRD neg vs. (VRd) 73% ≥VGPR 42% ≥CR of which 24% MRD neg | NA (FU 13.5 months) | >10% Cytopenia similar in two arms 40% of Dara infusion-related reaction (mainly G1–2) |
| NCT02541383 CASSIOPEIA [77] | Phase III | Dara-VTd-ASCT-Dara-VTd vs. VTd-ASCT-VTd Maintenance Daratumumab vs. observation | Induction (cycles 1–4) and consolidation (cycle 5–6) Bortezomib 1.3 mg/m2 D1, 4, 8, 11 Thalidomide 100 mg daily p.o. in all cycles Dexamethasone 40 mg p.o. or iv D1, 2, 8, 9, 15, 16, 22, and 23 of induction cycles 1 and 2 and D1 and 2 of induction cycles 3 and 4 and 20 mg on D8, 9, 15, 16 of induction cycles 3–4 and D1, 2, 8, 9, 15, and 16 of both consolidation cycles. Daratumumab iv 16 mg/kg once weekly in induction cycles 1 and 2 and once every 2 weeks during induction cycles 3 and 4 and consolidation Stem-cell mobilization with cyclophosphamide (3 g/m2) ASCT Mel200 mg/m2 iv conditioning Maintenance Daratumumab (16 mg/kg) every 8 weeks until disease progression or for a maximum of 2 years vs. observation | 1085 | Post-consolidation (Dara–VTd) 83% ≥VGPR 10% CR 28.9% sCR 64% MRD neg (MFC) 39% MRD neg (NGS) vs. (VTd) 78% ≥VGPR 6% CR 20.3% sCR 44% MRD neg (MFC) 23% MRD neg (NGS) | Probability: 18 months PFS (Dara–VTd) 93% vs. (VTd) 85% OS NR | Neutropenia 28% vs. 15% Thrombocytopenia 11 vs. 7% Neuropathy 9% vs. 9% GIT 16% Reaction infusion 4% in Dara–VTd |
| NCT01998971 MMY1001 [11] | Phase Ib | Dara-KRd | Daratumumab 16 mg/kg QW for cycles 1–2, Q2W for cycles 3-6, and Q4W (1st dose of Dara split over 2 days) Carfilzomib 20/36 mg/m2 iv weekly D1, 8 and 15 of each 28-day cycle (20 mg/m2 on cycle 1, D1) for ≤13 cycles or elective discontinuation for ASCT Lenalidomide p.o. 25 mg D1–21 and Dexamethasone 20–40 mg per week | 22 | Best response 33% VGPR 14% CR 43% sCR | 1-year PFS 95% | Lymphopenia 50% Neutropenia 23% 1 (5%) cardiac grade 3 TEAE was observed (congestive heart failure) |
| NCT01217957 C16005 [78] | Phase I/II | Ixa-Rd induction ASCT ± Ixa maintenance | Induction 28-day cycles (cycles 1–12) Ixazomib p.o. 4 mg D1, 8, and 15 Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg D1, 8, 15, and 22 Discontinuation → ASCT Maintenance Only for patients who did not proceed to ASCT: Single-agent ixazomib, given at the last tolerated dose during induction | 65 | 63% ≥VGPR 32% CR MRD neg 12.5% (MFC) | Median PFS 35.4 months | Neutropenia 14% Thrombocytopenia 9% GIT 6% |
| Protocol | Phase | Treatment | Subjects | Response | TTP/PFS/OS | Toxicity (≥G3) | |
|---|---|---|---|---|---|---|---|
| NCT00644228 SWOG S0777 [108] | Phase III | VRd vs. Rd | VRd 21-day cycles (cycles 1–8) Bortezomib 1.3 mg/m2 iv D1, 4, 8, and 11 +; Lenalidomide p.o. 25 mg daily D1–14 Dexamethasone p.o. 20 mg daily D1, 2, 4, 5, 8, 9, 11, and 12 Rd 28-day cycles (cycles 1–6) Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg D1, 8, 15, and 22 | 525 | ≥VGPR 43% vs. 32% CR 15.7% vs. 8.4% | Median PFS 43 vs/ 30 months, median OS 75 vs. 64 months | Neurological AEs 33% vs. 11% |
| NCT02195479 ALCYONE [27] | Phase III | Dara-VMp vs. VMp | Induction 42-day cycles (cycles 1–9) Bortezomib 1.3 mg/m2 sc twice-weekly on weeks 1, 2, 4, and 5 of cycle 1 and once weekly on weeks 1, 2, 4, and 5 of cycles 2–9) Melphalan p.o. 9 mg/m2 once-daily on D1–4 Prednisone p.o. 60 mg/m2, once-daily on D1–4 Daratumumab iv 16 mg/kg Dexamethasone 20 mg once-weekly in cycle 1, every 3 weeks in cycles 2–9, and every 4 weeks thereafter until disease progression | 706 | ≥VGPR 71% vs. 49% ≥CR 42% vs. 24% MRD neg by NGS Dara-VMp arm: 22.3% VMp arm: 6.2% | Median PFS NR | Neutropenia 40% vs. 38% Anemia 16% vs. 19%, Thrombocytopenia 34% vs. 37% Pneumonia 11% vs. 4% |
| NCT02252172 MAIA [110] | Phase III | Dara-Rd vs. Rd | Dara–Rd 28-day cycles Daratumumab iv 16 mg /kg once-weekly during cycles 1–2, every 2 weeks during cycles 3–6, and every 4 weeks thereafter Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg D1, 8, 15, and 22 Rd 28-day cycle Lenalidomide p.o. 25 mg D1–21 Dexamethasone p.o. 40 mg D1, 8, 15, and 22 | 737 | ≥VGPR 79% vs. 53% ≥CR 47% vs. 24% MRD neg by NGS Dara–Rd arm: 24.2% Rd arm: 7.3% | Median PFS NR | Neutropenia 50% vs. 35% Anemia 12% vs. 20% Lymphopenia 15% vs. 11% Pneumonia 14% vs. 8% |
| NCT02513186 SARVRD [111] | Phase I | Isa-VRd induction + Isa-Rd maintenance (16) | Induction 6-week cycles Isatuximab iv 10 mg/kg (cycles 1–4) D1, 8, 15, 22, 29 (cycle 1),; D1, 15, 29 (cycles 2–32 (cycles 1–4) Lenalidomide p.o. 25 mg/day D1–14 and D22–35 (cycles 1–4) Dexamethasone 20 mg D1, 2, 4, 5, 8, 9, 11, 12, 15, 22, 23, 25, 26, 29, 30, 32, 33 Maintenance 4-week cycles Isatuximab iv 10 mg/kg on D1, 15 (all cycles) Lenalidomide p.o. 25 mg D1–21 (all cycles) Dexamethasone p.o. 40 mg D1, 8, 15, 22 (all cycles) | 22 | ORR 93% MRD neg by NGS 50% (33% at 10−6) NGF 44% (18% at 10−6) | NA | G≥3 AEs were reported in 10 (46%) and SAEs in 4 (18%) pts Lymphopenia (8/22) Neutropenia (4/22) Thrombocytopenia (4/22) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D'Agostino, M.; Bertamini, L.; Oliva, S.; Boccadoro, M.; Gay, F. Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies. Cancers 2019, 11, 2015. https://doi.org/10.3390/cancers11122015
D'Agostino M, Bertamini L, Oliva S, Boccadoro M, Gay F. Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies. Cancers. 2019; 11(12):2015. https://doi.org/10.3390/cancers11122015
Chicago/Turabian StyleD'Agostino, Mattia, Luca Bertamini, Stefania Oliva, Mario Boccadoro, and Francesca Gay. 2019. "Pursuing a Curative Approach in Multiple Myeloma: A Review of New Therapeutic Strategies" Cancers 11, no. 12: 2015. https://doi.org/10.3390/cancers11122015