HuR Reduces Radiation-Induced DNA Damage by Enhancing Expression of ARID1A

 , , , , , and
, , , , , and 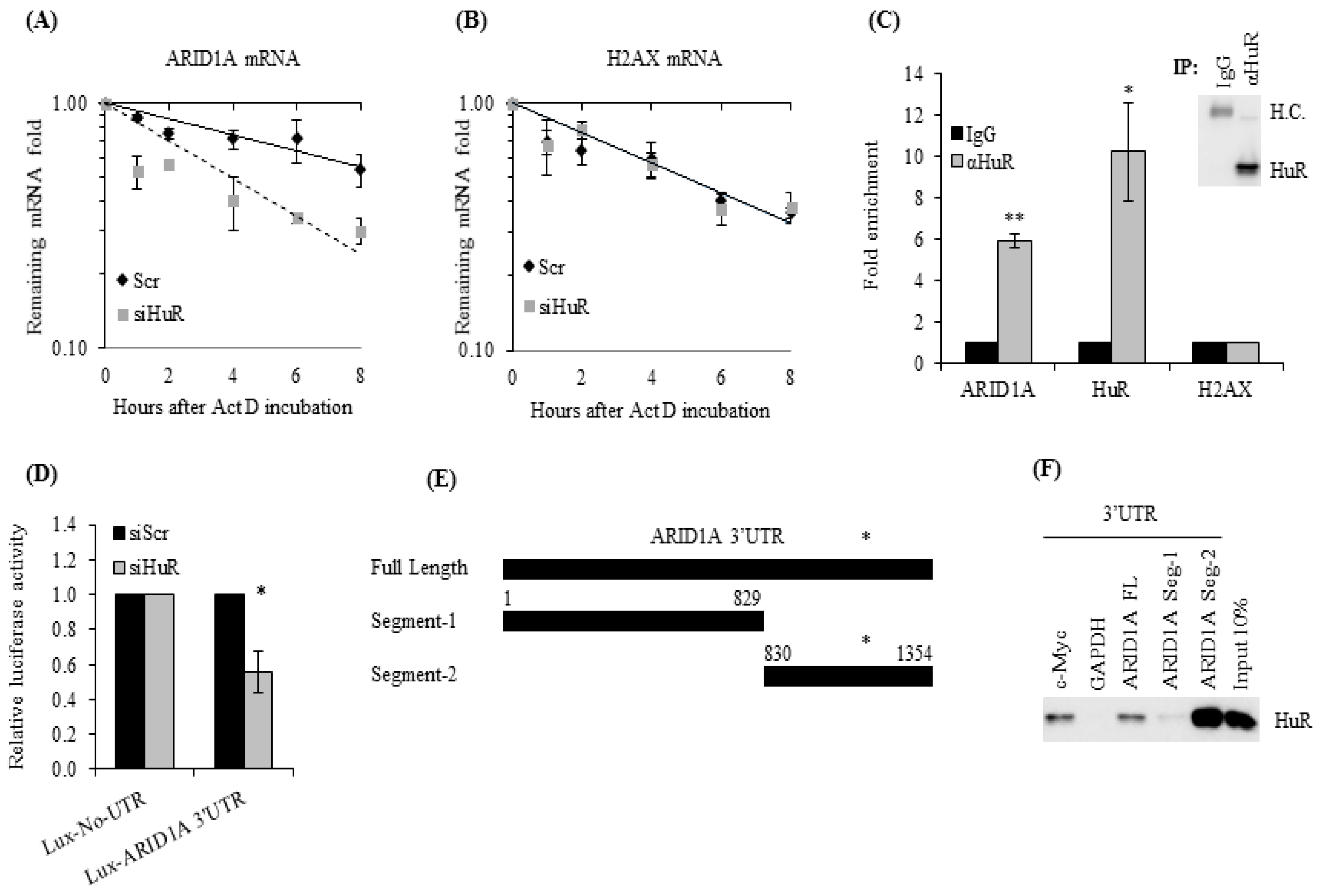{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ARID1A mRNA Is a Novel HuR Target
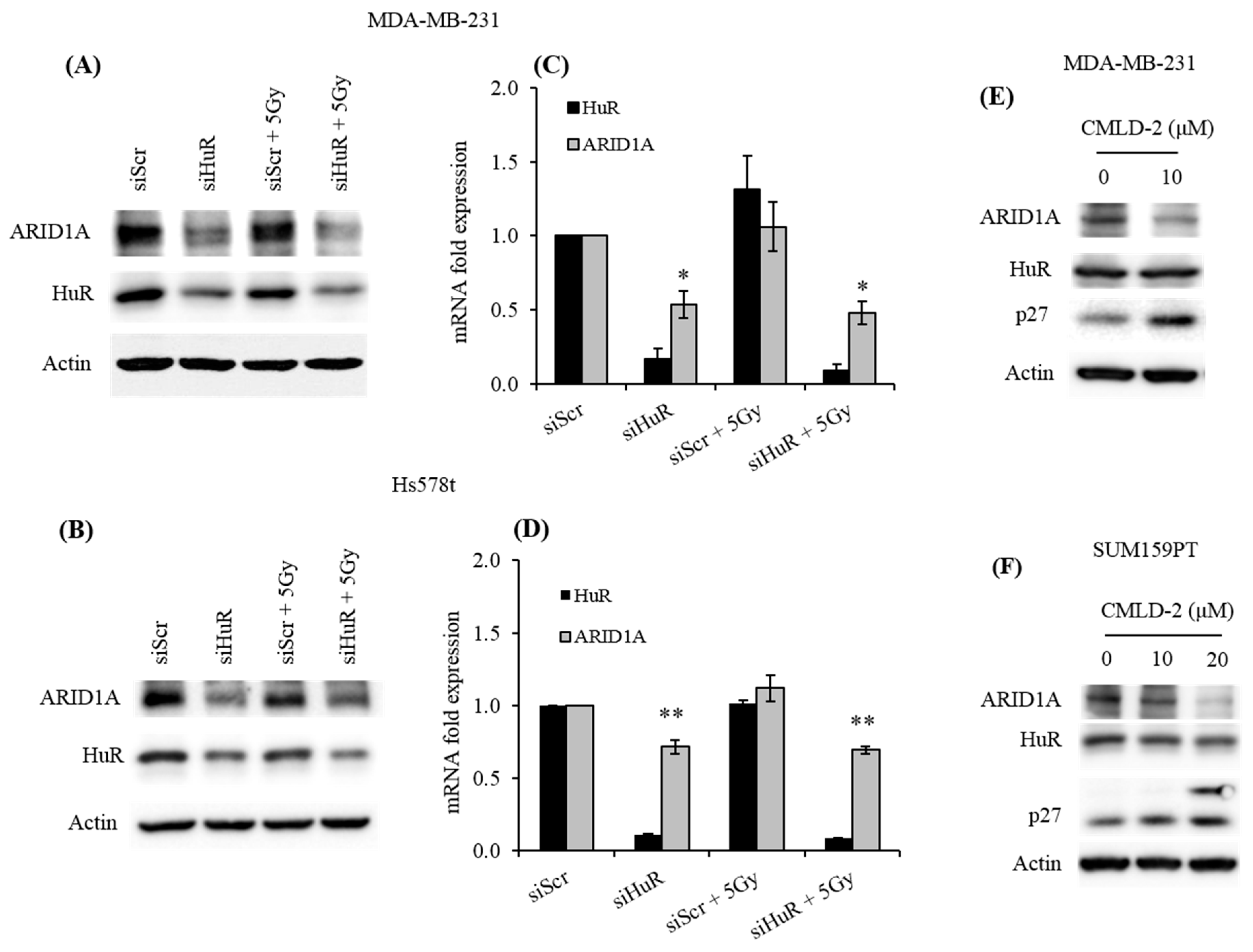
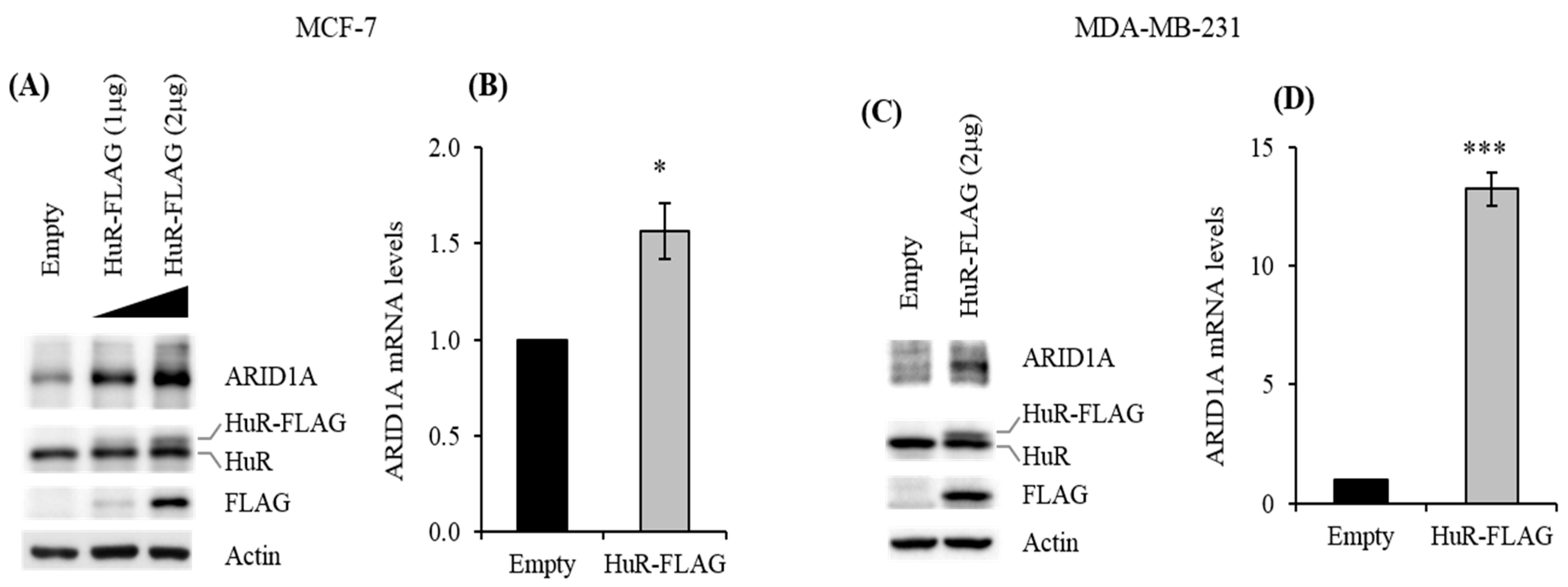
2.2. HuR Promotes ARID1A Expression
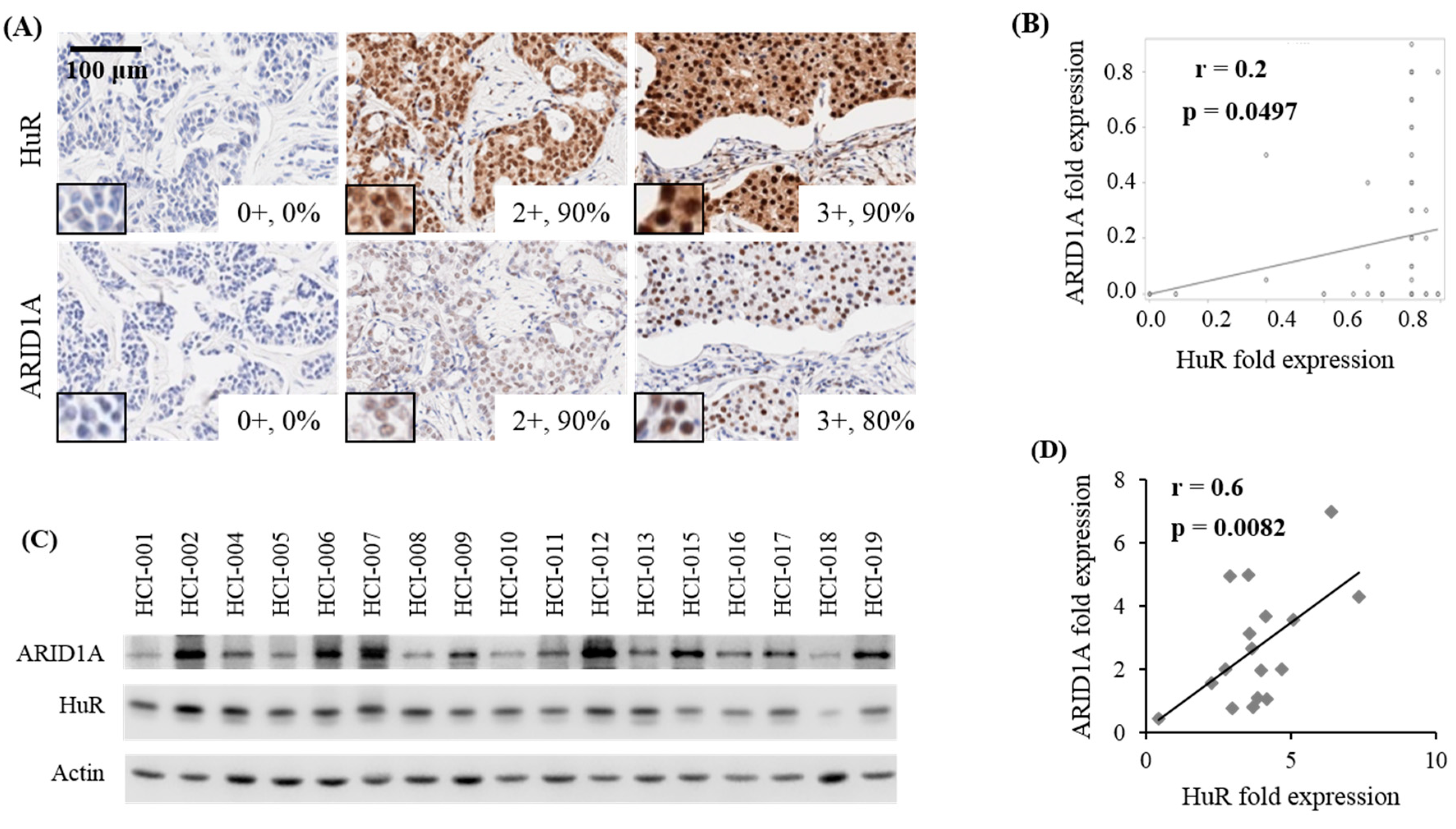
2.3. Correlation Between HuR and ARID1A Expression in Tumors of Patients with Breast Cancer
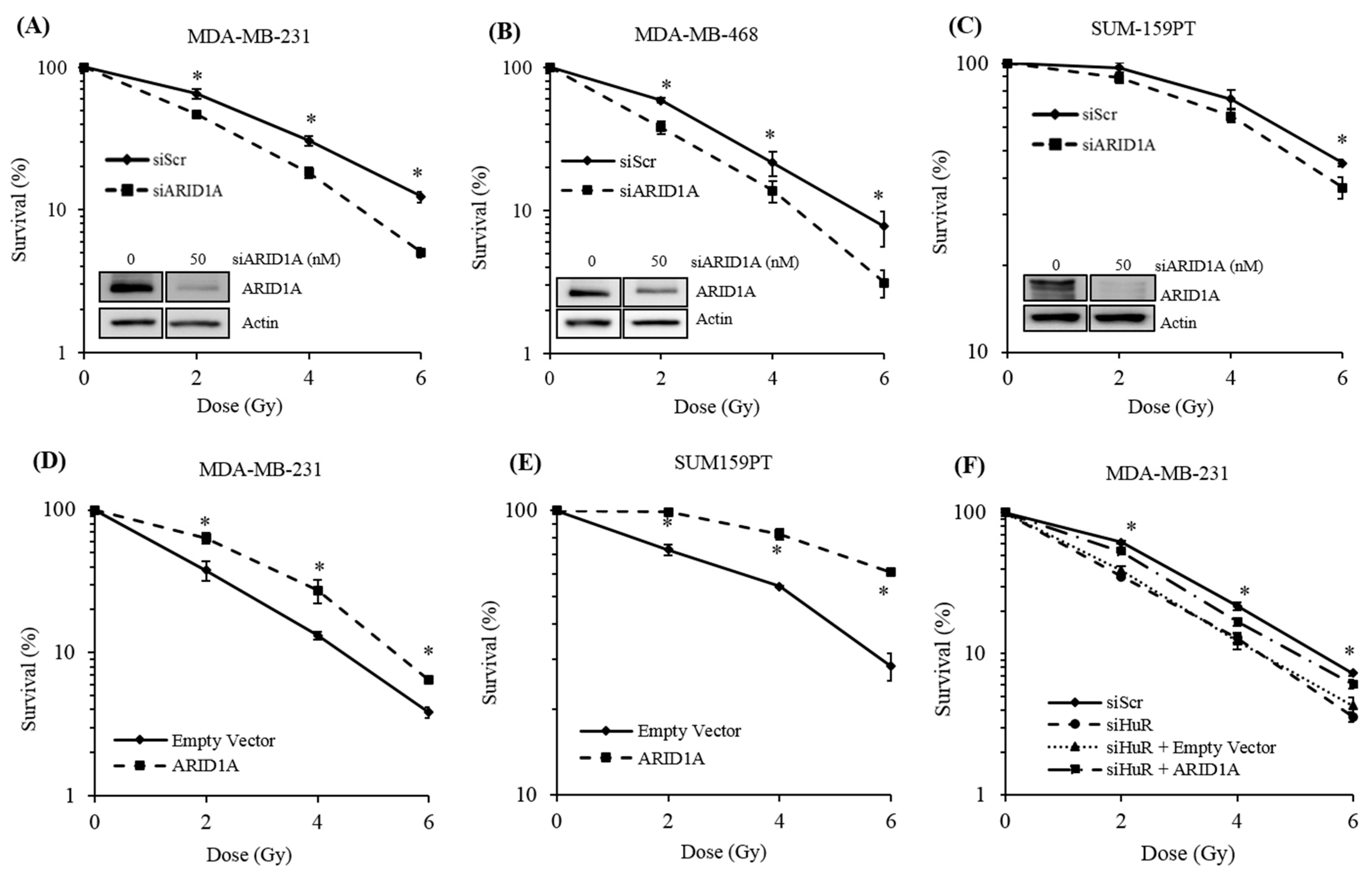
2.4. Genetic Inhibition of ARID1A Radiosensitizes Breast Cancer Cell Lines
2.5. ARID1A Expression Complements HuR Inhibition During Resistance to Radiation
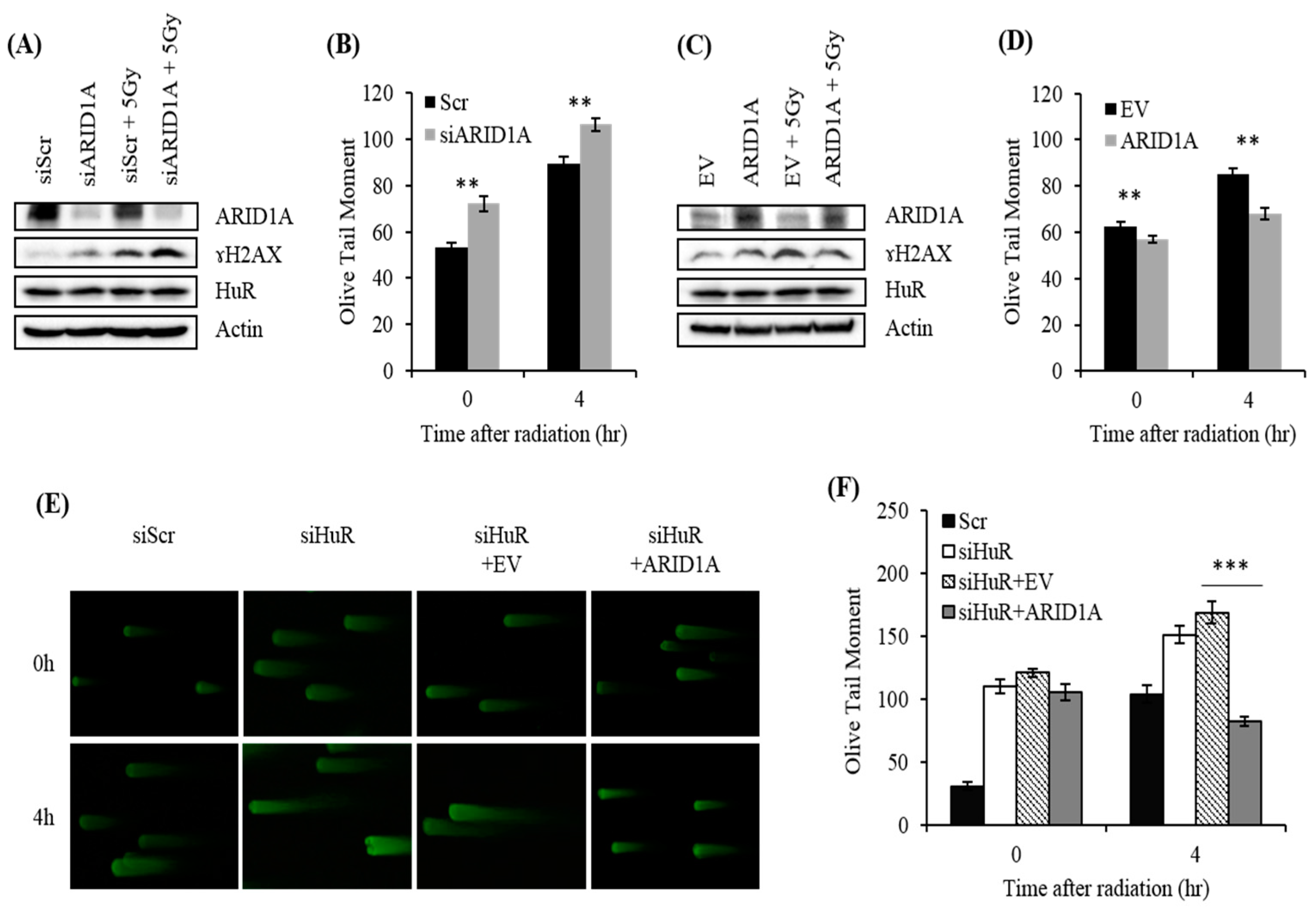
2.6. Impact of ARID1A on Radiation-Induced DSB in HuR-Inhibited Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Reagents
4.3. PDX Tissue Samples and Tissue Arrays
4.4. Transient Transfection
4.5. RT-qPCR
4.6. Western Blotting
4.7. Ribonucleoprotein Immunoprecipitation (RIP) Analysis
4.8. Luciferase Assay
4.9. Analysis of ARID1A mRNA Stability in HuR-Silenced Cells
4.10. Exposure to Ionizing Radiation
4.11. Clonogenic Assay
4.12. Biotinylated RNA Pulldown
4.13. Data Mining
4.14. Comet Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López de Silanes, I.; Zhan, M.; Lal, A.; Yang, X.; Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2987–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Furneaux, H.; Cheng, H.; Caldwell, M.C.; Hutter, D.; Liu, Y.; Holbrook, N.; Gorospe, M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000, 20, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.H.; Abdelmohsen, K.; Gorospe, M. Regulation of HuR by DNA Damage Response Kinases. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, S.; Burkhart, R.A.; Beeharry, N.; Bhattacharjee, V.; Londin, E.R.; Cozzitorto, J.A.; Romeo, C.; Jimbo, M.; Norris, Z.A.; Yeo, C.J.; et al. HuR post-transcriptionally regulates WEE1: Implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014, 74, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Pineda, D.M.; Rittenhouse, D.W.; Valley, C.C.; Cozzitorto, J.A.; Burkhart, R.A.; Leiby, B.; Winter, J.M.; Weber, M.C.; Londin, E.R.; Rigoutsos, I.; et al. HuR’s post-transcriptional regulation of death receptor 5 in pancreatic cancer cells. Cancer. Biol. Ther. 2012, 13, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Abdelmohsen, K.; Lal, A.; Kim, H.H.; Gorospe, M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef]
- López de Silanes, I.; Fan, J.; Yang, X.; Zonderman, A.B.; Potapova, O.; Pizer, E.S.; Gorospe, M. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003, 22, 7146–7154. [Google Scholar] [CrossRef]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cell. Mol. Life. Sci. 2008, 65, 3168–3181. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, B.; Bi, J.; Zhang, C. Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med. Oncol. 2011, 28, S577–S585. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Kuwano, Y.; Kim, H.H.; Gorospe, M. Posttranscriptional gene regulation by RNA binding proteins during oxidative stress: Implications for cellular senescence. Biol. Chem. 2008, 389, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Bono, P.; Narko, K.; Chang, S.H.; Lundin, J.; Joensuu, H.; Furneaux, H.; Hla, T.; Haglund, C.; Ristimäki, A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005, 65, 2157–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippova, N.; Yang, X.; Wang, Y.; Gillespie, G.Y.; Langford, C.; King, P.H.; Wheeler, C.; Nabors, L.B. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer. Res. 2011, 9, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetter, C.; Licata, L.A.; Witkiewicz, A.; Costantino, C.L.; Yeo, C.J.; Brody, J.R.; Keen, J.C. Cytoplasmic accumulation of the RNA binding protein HuR is central to tamoxifen resistance in estrogen receptor positive breast cancer cells. Cancer Biol. Ther. 2008, 7, 1496–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chang, R.; Ji, W.; Wang, N.; Qi, M.; Xu, Y.; Guo, J.; Zhan, L. Loss of Scribble Promotes Snail Translation through Translocation of HuR and Enhances Cancer Drug Resistance. J. Biol. Chem. 2016, 291, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.K.; Leung, W.W.; Ng, S.S. Exploiting a novel miR-519c-HuR-ABCG2 regulatory pathway to overcome chemoresistance in colorectal cancer. Exp. Cell. Res. 2015, 338, 222–231. [Google Scholar] [CrossRef]
- Mehta, M.; Basalingappa, K.; Griffith, J.N.; Andrade, D.; Babu, A.; Amreddy, N.; Muralidharan, R.; Gorospe, M.; Herman, T.; Ding, W.Q.; et al. HuR silencing elicits oxidative stress and DNA damage and sensitizes human triple-negative breast cancer cells to radiotherapy. Oncotarget 2016, 7, 64820–64835. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodelers and cancer. Nat. Rev. Cancer. 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Kaeser, M.D.; Aslanian, A.; Dong, M.Q.; Yates, J.R., 3rd; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 2008, 283, 32254–32263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Chambers, K.J.; Faller, D.V.; Wang, S. Reprogramming of the SWI/SNF complex for co-activation or co-repression in prohibitin-mediated estrogen receptor regulation. Oncogene 2007, 26, 7153–7157. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenet. Chromatin. 2012, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeenk, G.; van Attikum, H. The chromatin response to DNA breaks: Leaving a mark on genome integrity. Ann. Rev. Biochem. 2013, 82, 55–80. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Seeber, A.; Hauer, M.; Gasser, S.M. Nucleosome remodelers in double-strand break repair. Curr. Opin. Genet. Dev. 2013, 23, 174–184. [Google Scholar] [CrossRef]
- Osley, M.A.; Tsukuda, T.; Nickoloff, J.A. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat. Res. 2007, 618, 65–80. [Google Scholar] [CrossRef]
- Chai, B.; Huang, J.; Cairns, B.R.; Laurent, B.C. Distinct roles for the RSC and SWI/SNF ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 2005, 19, 1656–1661. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Kothandapani, A.; Gopalakrishnan, K.; Kahali, B.; Reisman, D.; Patrick, S.M. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp. Cell Res. 2012, 318, 1973–1986. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.H.; Chakraborty, A.R.; Mo, X.; Liu, Z.; Shilo, K.; Kirste, S.; Stegmaier, P.; McNulty, M.; Karachaliou, N.; Rosell, R.; et al. SMARCA4/BRG1 Is a Novel Prognostic Biomarker Predictive of Cisplatin-Based Chemotherapy Outcomes in Resected Non-Small Cell Lung Cancer. Clin. Cancer. Res. 2016, 22, 2396–2404. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisman, D.; Glaros, S.; Thompson, E.A. The SWI/SNF complex and cancer. Oncogene 2009, 28, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoch, C.; Crabtree, G.R. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Nat. Rev. Cancer. 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.I.; Lessard, J.; Crabtree, G.R. Understanding the words of chromatin regulation. Cell 2009, 136, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Nagl, N.G., Jr.; Patsialou, A.; Haines, D.S.; Dallas, P.B.; Beck, G.R., Jr.; Moran, E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005, 65, 9236–9244. [Google Scholar] [CrossRef] [Green Version]
- Allo, G.; Bernardini, M.Q.; Wu, R.C.; Shih, I.M.; Kalloger, S.; Pollett, A.; Gilks, C.B.; Clarke, B.A. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Mod. Pathol. 2014, 27, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Li, J.H.; Shao, P.; Zhou, H.; Chen, Y.Q.; Qu, L.H. starBase: A database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011, 39, D202–D209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; Tunge, J.A.; et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, M.; Göpfert, U.; Siewe, B.; Hengst, L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5’UTR. Genes Dev. 2002, 16, 3087–3099. [Google Scholar] [CrossRef] [Green Version]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Tabchy, A.; Valero, V.; Vidaurre, T.; Lluch, A.; Gomez, H.; Martin, M.; Qi, Y.; Barajas-Figueroa, L.J.; Souchon, E.; Coutant, C.; et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin. Cancer Res. 2010, 16, 5351–5361. [Google Scholar] [CrossRef] [Green Version]
- Stickeler, E.; Pils, D.; Klar, M.; Orlowsk-Volk, M.; Zur Hausen, A.; Jäger, M.; Watermann, D.; Gitsch, G.; Zeillinger, R.; Tempfer, C.B. Basal-like molecular subtype and HER4 up-regulation and response to neoadjuvant chemotherapy in breast cancer. Oncol. Rep. 2011, 26, 1037–1045. [Google Scholar]
- Zhang, X.; Zou, T.; Rao, J.; Liu, L.; Xiao, L.; Wang, P.Y.; Cui, Y.H.; Gorospe, M.; Wang, J.Y. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res. 2009, 37, 7623–7637. [Google Scholar] [CrossRef]
- Laroia, G.; Cuesta, R.; Brewer, G.; Schneider, R.J. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science 1999, 284, 499–502. [Google Scholar] [CrossRef]
- Xiao, L.; Rao, J.N.; Zou, T.; Liu, L.; Marasa, B.S.; Chen, J.; Turner, D.J.; Zhou, H.; Gorospe, M.; Wang, J.Y. Polyamines regulate the stability of activating transcription factor-2 mRNA through RNA-binding protein HuR in intestinal epithelial cells. Mol. Biol. Cell 2007, 18, 4579–4590. [Google Scholar] [CrossRef]
- Zou, T.; Mazan-Mamczarz, K.; Rao, J.N.; Liu, L.; Marasa, B.S.; Zhang, A.H.; Xiao, L.; Pullmann, R.; Gorospe, M.; Wang, J.Y. Polyamine depletion increases cytoplasmic levels of RNA binding protein HuR leading to stabilization of nucleophosmin and p53 mRNAs. J. Biol. Chem. 2006, 281, 19387–19389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwano, Y.; Kim, H.H.; Abdelmohsen, K.; Pullmann, R., Jr.; Martindale, J.L.; Yang, X.; Gorospe, M. MKP-1 mRNA stabilization and translational control by RNA-binding proteins HuR and NF90. Mol. Cell. Biol. 2008, 28, 4562–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, B.; Wang, T.L.; Shih, I.M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, R.; Mehta, M.; Ahmed, R.; Roy, S.; Xu, L.; Aubé, J.; Chen, A.; Zhao, Y.D.; Herman, T.; Ramesh, R.; et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci. Rep. 2017, 7, 9694–9704. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Gligorich, K.M.; Wang, G.; Georgelas, A.; Bowman, P.; Courdy, S.J.; Welm, A.L.; Welm, B.E. Patient-derived models of human breast cancer: Protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr. Protoc. Pharmacol. 2013, 60, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Prot. 2006, 1, 302–307. [Google Scholar] [CrossRef]
- Paschoud, S.; Dogar, A.M.; Kuntz, C.; Grisoni-Neupert, B.; Richman, L.; Kühn, L.C. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell. Biol. 2006, 26, 8228–8241. [Google Scholar] [CrossRef] [Green Version]
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-transcriptional regulation of programmed cell death 4 (PDCD4) mRNA by the RNA-binding proteins human antigen R (HuR) and T-cell intracellular antigen 1 (TIA1). J. Biol. Chem. 2015, 290, 3468–3487. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade, D.; Mehta, M.; Griffith, J.; Oh, S.; Corbin, J.; Babu, A.; De, S.; Chen, A.; Zhao, Y.D.; Husain, S.; et al. HuR Reduces Radiation-Induced DNA Damage by Enhancing Expression of ARID1A. Cancers 2019, 11, 2014. https://doi.org/10.3390/cancers11122014
Andrade D, Mehta M, Griffith J, Oh S, Corbin J, Babu A, De S, Chen A, Zhao YD, Husain S, et al. HuR Reduces Radiation-Induced DNA Damage by Enhancing Expression of ARID1A. Cancers. 2019; 11(12):2014. https://doi.org/10.3390/cancers11122014
Chicago/Turabian StyleAndrade, Daniel, Meghna Mehta, James Griffith, Sangphil Oh, Joshua Corbin, Anish Babu, Supriyo De, Allshine Chen, Yan D. Zhao, Sanam Husain, and et al. 2019. "HuR Reduces Radiation-Induced DNA Damage by Enhancing Expression of ARID1A" Cancers 11, no. 12: 2014. https://doi.org/10.3390/cancers11122014