Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development



Abstract
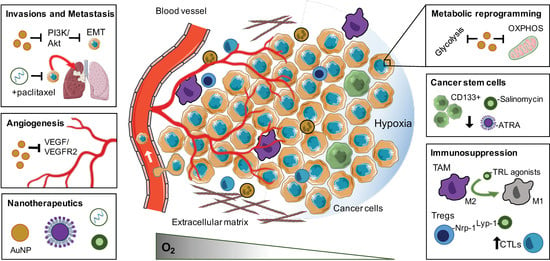
:
1. Introduction
1.1. Defining Tumour Hypoxia
1.2. Implications of Tumour Hypoxia and Nanotherapeutic Opportunities
1.3. Scope of the Review
2. Biological Response and Therapeutic Opportunities of Tumour Hypoxia
2.1. Enrichment and Propagation of Cancer Stem Cells
Nanotherapeutic Approaches to Target Cancer Stem Cells
2.2. Invasions and Metastasis
Targeting EMT and Metastatic Progression with Nanoparticle Formulations
2.3. Angiogenesis
Overcoming Hypoxia-Driven Angiogenesis Using Nanoparticles
2.4. Immunosuppression
Reprogramming the Immunosuppressive TME with Nanotherapeutics
2.5. Metabolic Reprogramming
Nanotherapeutics to Target or Overcome Metabolic Reprogramming
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Horsman, M.R.; Vaupel, P. Pathophysiological basis for the formation of the tumor microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, M.W. Concepts of oxygen transport at the microcirculatory level. In Seminars in Radiation Oncology; Elsevier: Amsterdam, The Netherlands, 1998; Volume 8, pp. 143–150. [Google Scholar]
- Durand, R.E.; Sham, E. The lifetime of hypoxic human tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 711–715. [Google Scholar] [CrossRef]
- Dubsky, P.; Sevelda, P.; Jakesz, R.; Hausmaninger, H.; Samonigg, H.; Seifert, M.; Denison, U.; Mlineritsch, B.; Steger, G.; Kwasny, W. Anemia is a significant prognostic factor in local relapse-free survival of premenopausal primary breast cancer patients receiving adjuvant cyclophosphamide/methotrexate/5-fluorouracil chemotherapy. Clin. Cancer Res. 2008, 14, 2082–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Plavetić, D.; Plavetić, N.D.; Barić, M.B.; Bradić, L.B.; Kulić, A.N.A.; Pleština, S. Hypoxia in solid tumors: Biological responses to hypoxia and implications on therapy and prognosis. Period. Biol. 2014, 116, 361–364. [Google Scholar]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: New approaches for combinational immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Prasad, M.; Lambe, U.P.; Brar, B.; Shah, I.; Manimegalai, J.; Ranjan, K.; Rao, R.; Kumar, S.; Mahant, S.; Khurana, S.K.; et al. Nanotherapeutics: An insight into healthcare and multi-dimensional applications in medical sector of the modern world. Biomed. Pharmacother. 2018, 97, 1521–1537. [Google Scholar] [CrossRef]
- Thakor, A.S.; Jokerst, J.V.; Ghanouni, P.; Campbell, J.L.; Mittra, E.; Gambhir, S.S. Clinically Approved Nanoparticle Imaging Agents. J. Nucl. Med. 2016, 57, 1833–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Province, P.; Griguer, C.E.; Han, X.; Shaykh, H.F. Hypoxia, angiogenesis and mechanisms for invasion of malignant gliomas. In Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications; IntechOpen: London, UK, 2013. [Google Scholar]
- Qiu, G.-Z.; Jin, M.-Z.; Dai, J.-X.; Sun, W.; Feng, J.-H.; Jin, W.-L. Reprogramming of the tumor in the hypoxic niche: The emerging concept and associated therapeutic strategies. Trends Pharmacol. Sci. 2017, 38, 669–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q.; Yu, Z. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar]
- Mi, Y.; Huang, Y.; Deng, J. The enhanced delivery of salinomycin to CD133+ ovarian cancer stem cells through CD133 antibody conjugation with poly (lactic-co-glycolic acid)-poly (ethylene glycol) nanoparticles. Oncol. Lett. 2018, 15, 6611–6621. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zeng, Y.; Qi, X.; Chen, Y.; Ge, Z.; Jiang, Z.; Zhang, X.; Dong, Y.; Chen, H.; Yu, Z. Targeted salinomycin delivery with EGFR and CD133 aptamers based dual-ligand lipid-polymer nanoparticles to both osteosarcoma cells and cancer stem cells. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2115–2127. [Google Scholar] [CrossRef]
- Gui, K.; Zhang, X.; Chen, F.; Ge, Z.; Zhang, S.; Qi, X.; Sun, J.; Yu, Z. Lipid-polymer nanoparticles with CD133 aptamers for targeted delivery of all-trans retinoic acid to osteosarcoma initiating cells. Biomed. Pharmacother. 2019, 111, 751–764. [Google Scholar] [CrossRef]
- Kesharwani, P.; Banerjee, S.; Padhye, S.; Sarkar, F.H.; Iyer, A.K. Hyaluronic acid engineered nanomicelles loaded with 3, 4-difluorobenzylidene curcumin for targeted killing of CD44+ stem-like pancreatic cancer cells. Biomacromolecules 2015, 16, 3042–3053. [Google Scholar] [CrossRef]
- Rao, W.; Wang, H.; Han, J.; Zhao, S.; Dumbleton, J.; Agarwal, P.; Zhang, W.; Zhao, G.; Yu, J.; Zynger, D.L.; et al. Chitosan-decorated doxorubicin-encapsulated nanoparticle targets and eliminates tumor reinitiating cancer stem-like cells. ACS Nano 2015, 9, 5725–5740. [Google Scholar] [CrossRef]
- Gong, Z.; Chen, D.; Xie, F.; Liu, J.; Zhang, H.; Zou, H.; Yu, Y.; Chen, Y.; Sun, Z.; Wang, X.; et al. Codelivery of salinomycin and doxorubicin using nanoliposomes for targeting both liver cancer cells and cancer stem cells. Nanomedicine 2016, 11, 2565–2579. [Google Scholar] [CrossRef]
- Muntimadugu, E.; Kumar, R.; Saladi, S.; Rafeeqi, T.A.; Khan, W. CD44 targeted chemotherapy for co-eradication of breast cancer stem cells and cancer cells using polymeric nanoparticles of salinomycin and paclitaxel. Colloids Surf. B Biointerfaces 2016, 143, 532–546. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Y.; Li, S.-Y.; Shen, S.; Du, X.-J.; Xu, C.-F.; Cao, Z.-T.; Bao, Y.; Zhu, Y.-H.; Li, Y.-P.; et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials 2015, 37, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yu, D.; Jia, B.; Jin, C.; Liu, X.; Zhao, X.; Zhang, G. Targeting CD133+ laryngeal carcinoma cells with chemotherapeutic drugs and siRNA against ABCG2 mediated by thermo/pH-sensitive mesoporous silica nanoparticles. Tumor Biol. 2016, 37, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mukherjee, P.; Chatterjee, R.; Jamal, Z.; Chatterji, U. Enhancing Chemosensitivity of Breast Cancer Stem Cells by Downregulating SOX2 and ABCG2 Using Wedelolactone-encapsulated Nanoparticles. Mol. Cancer Ther. 2019, 18, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Mamaeva, V.; Niemi, R.; Beck, M.; Ozliseli, E.; Desai, D.; Landor, S.; Gronroos, T.; Kronqvist, P.; Pettersen, I.K.; McCormack, E.; et al. Inhibiting Notch Activity in Breast Cancer Stem Cells by Glucose Functionalized Nanoparticles Carrying gamma-secretase Inhibitors. Mol. Ther. 2016, 24, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Saha, S.; Wang, E.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Inhibition of tumor growth and metastasis by a self-therapeutic nanoparticle. Proc. Natl. Acad. Sci. USA 2013, 110, 6700–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulens-Arias, V.; Rojas, J.M.; Pérez-Yagüe, S.; del Puerto Morales, M.; Barber, D.F. Polyethylenimine-coated SPION exhibits potential intrinsic anti-metastatic properties inhibiting migration and invasion of pancreatic tumor cells. J. Control. Release 2015, 216, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Wu, Z.; Wang, J.; Li, Q.; Shen, S.; Wang, W.; Zhou, L.; Wang, W.; Cao, Z.; Guo, Y. Development of a Non-Coding-RNA-based EMT/CSC Inhibitory Nanomedicine for In Vivo Treatment and Monitoring of HCC. Adv. Sci. 2019, 6, 1801885. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zhou, B.; Luo, H.; Mao, J.; Huang, Y.; Zhang, K.; Mei, C.; Yan, Y.; Jin, H.; Gao, J. ZnAs@ SiO2 nanoparticles as a potential anti-tumor drug for targeting stemness and epithelial-mesenchymal transition in hepatocellular carcinoma via SHP-1/JAK2/STAT3 signaling. Theranostics 2019, 9, 4391. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Yoo, K.C.; Uddin, N.; Kim, J.S.; Lee, S.J.; Choi, E.H. Low doses of PEG-coated gold nanoparticles sensitize solid tumors to cold plasma by blocking the PI3K/AKT-driven signaling axis to suppress cellular transformation by inhibiting growth and EMT. Biomaterials 2016, 87, 118–130. [Google Scholar] [CrossRef]
- Tang, S.; Yin, Q.; Su, J.; Sun, H.; Meng, Q.; Chen, Y.; Chen, L.; Huang, Y.; Gu, W.; Xu, M.; et al. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (beta-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials 2015, 48, 1–15. [Google Scholar] [CrossRef]
- Shahin, S.A.; Wang, R.; Simargi, S.I.; Contreras, A.; Parra Echavarria, L.; Qu, L.; Wen, W.; Dellinger, T.; Unternaehrer, J.; Tamanoi, F.; et al. Hyaluronic acid conjugated nanoparticle delivery of siRNA against TWIST reduces tumor burden and enhances sensitivity to cisplatin in ovarian cancer. Nanomedicine 2018, 14, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Bhattacharya, R.; Wang, P.; Wang, L.; Basu, S.; Nagy, J.A.; Atala, A.; Mukhopadhyay, D.; Soker, S. Antiangiogenic properties of gold nanoparticles. Clin. Cancer Res. 2005, 11, 3530–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, X.; Liu, S.; Yang, W.; Pan, F.; Yang, X.Y.; Du, B.; Qin, L.; Pan, Y. Gold nanoparticles attenuate metastasis by tumor vasculature normalization and epithelial-mesenchymal transition inhibition. Int. J. Nanomed. 2017, 12, 3509–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, F.; Li, W.; Yang, W.; Yang, X.Y.; Liu, S.; Li, X.; Zhao, X.; Ding, H.; Qin, L.; Pan, Y. Anterior gradient 2 as a supervisory marker for tumor vessel normalization induced by anti-angiogenic treatment. Oncol. Lett. 2018, 16, 3083–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunjachan, S.; Detappe, A.; Kumar, R.; Ireland, T.; Cameron, L.; Biancur, D.E.; Motto-Ros, V.; Sancey, L.; Sridhar, S.; Makrigiorgos, G.M.; et al. Nanoparticle mediated tumor vascular disruption: A novel strategy in radiation therapy. Nano Lett. 2015, 15, 7488–7496. [Google Scholar] [CrossRef]
- Wu, P.; Onodera, Y.; Ichikawa, Y.; Rankin, E.B.; Giaccia, A.J.; Watanabe, Y.; Qian, W.; Hashimoto, T.; Shirato, H.; Nam, J.-M. Targeting integrins with RGD-conjugated gold nanoparticles in radiotherapy decreases the invasive activity of breast cancer cells. Int. J. Nanomed. 2017, 12, 5069–5085. [Google Scholar] [CrossRef] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Han, S.; Wang, W.; Wang, S.; Wang, S.; Ju, R.; Pan, Z.; Yang, T.; Zhang, G.; Wang, H.; Wang, L. Multifunctional biomimetic nanoparticles loading baicalin for polarizing tumor-associated macrophages. Nanoscale 2019, 11, 20206–20220. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, L.; Liu, Q.; Hou, L.; Huang, L. Inhibiting PI3 kinase-γ in both myeloid and plasma cells remodels the suppressive tumor microenvironment in desmoplastic tumors. J. Control. Release 2019, 309, 173–180. [Google Scholar] [CrossRef]
- Wu, C.; Muroski, M.E.; Miska, J.; Lee-Chang, C.; Shen, Y.; Rashidi, A.; Zhang, P.; Xiao, T.; Han, Y.; Lopez-Rosas, A.; et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 126–137. [Google Scholar] [CrossRef]
- Ou, W.; Thapa, R.K.; Jiang, L.; Soe, Z.C.; Gautam, M.; Chang, J.H.; Jeong, J.H.; Ku, S.K.; Choi, H.G.; Yong, C.S.; et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J. Control. Release 2018, 281, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, T.; Yu, H.; Feng, B.; Zhou, L.; Zhou, F.; Hou, B.; Zhang, H.; Luo, M.; Li, Y. Engineering nanoparticles to locally activate T cells in the tumor microenvironment. Sci. Immunol. 2019, 4, eaau6584. [Google Scholar] [CrossRef] [PubMed]
- Marrache, S.; Dhar, S. The energy blocker inside the power house: Mitochondria targeted delivery of 3-bromopyruvate. Chem. Sci. 2015, 6, 1832–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Zhou, T.; Cui, P.; He, Y.; Chang, X.; Xing, L.; Jiang, H. Modulation of Intracellular Oxygen Pressure by Dual-Drug Nanoparticles to Enhance Photodynamic Therapy. Adv. Funct. Mater. 2019, 29, 1806708. [Google Scholar] [CrossRef]
- Xia, D.; Xu, P.; Luo, X.; Zhu, J.; Gu, H.; Huo, D.; Hu, Y. Overcoming Hypoxia by Multistage Nanoparticle Delivery System to Inhibit Mitochondrial Respiration for Photodynamic Therapy. Adv. Funct. Mater. 2019, 29, 1807294. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, J.; Liu, S.; Li, X.; Miao, L.; Yang, B.; Zhang, C.; He, J.; Ai, S.; Guan, W. Defeating relapsed and refractory malignancies through a nano-enabled mitochondria-mediated respiratory inhibition and damage pathway. Biomaterials 2019, 229, 119580. [Google Scholar] [CrossRef]
- Zuo, H.; Tao, J.; Shi, H.; He, J.; Zhou, Z.; Zhang, C. Platelet-mimicking nanoparticles co-loaded with W18O49 and metformin alleviate tumor hypoxia for enhanced photodynamic therapy and photothermal therapy. Acta Biomater. 2018, 80, 296–307. [Google Scholar] [CrossRef]
- Kang, N.; Choi, S.Y.; Kim, B.N.; Yeo, C.D.; Park, C.K.; Kim, Y.K.; Kim, T.-J.; Lee, S.-B.; Lee, S.H.; Park, J.Y.; et al. Hypoxia-induced cancer stemness acquisition is associated with CXCR4 activation by its aberrant promoter demethylation. BMC Cancer 2019, 19, 148. [Google Scholar] [CrossRef] [Green Version]
- Prasad, P.; Mittal, S.A.; Chongtham, J.; Mohanty, S.; Srivastava, T. Hypoxia-Mediated Epigenetic Regulation of Stemness in Brain Tumor Cells. Stem Cells 2017, 35, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhou, C.; Xu, L.; Xiao, H. Hypoxia enhances stemness of cancer stem cells in glioblastoma: An in vitro study. Int. J. Med. Sci. 2013, 10, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Lan, C.; Xiong, S.; Zhao, X.; Shan, Y.; Hu, R.; Wan, W.; Yu, S.; Liao, B.; Li, G.; et al. HIF1α regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia. Oncotarget 2017, 8, 28074. [Google Scholar] [PubMed] [Green Version]
- Jacobsson, H.; Harrison, H.; Hughes, É.; Persson, E.; Rhost, S.; Fitzpatrick, P.; Gustafsson, A.; Andersson, D.; Gregersson, P.; Magnusson, Y.; et al. Hypoxia-induced secretion stimulates breast cancer stem cell regulatory signalling pathways. Mol. Oncol. 2019, 13, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Asghari, F.; Khademi, R.; Esmaeili Ranjbar, F.; Veisi Malekshahi, Z.; Faridi Majidi, R. Application of Nanotechnology in Targeting of Cancer Stem Cells: A Review. Int. J. Stem Cells 2019, 12, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Boeuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef]
- Kim, D.; Choi, B.H.; Ryoo, I.G.; Kwak, M.K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: Inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018, 9, 896. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T. Targeting cancer stem cells by curcumin and clinical applications. Cancer Lett. 2014, 346, 197–205. [Google Scholar] [CrossRef]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.-R.; Falasca, M. ABC transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Chen, J.; Zhang, J.; Chen, H.; Yan, M.; Huang, L.; Tian, Y.; Chen, Y.; Wang, Y. Hypoxia induces TWIST-activated epithelial–mesenchymal transition and proliferation of pancreatic cancer cells in vitro and in nude mice. Cancer Lett. 2016, 383, 73–84. [Google Scholar] [CrossRef]
- Zhang, X.; Sai, B.; Wang, F.; Wang, L.; Wang, Y.; Zheng, L.; Li, G.; Tang, J.; Xiang, J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer 2019, 18, 40. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, H.; Wu, M.; Feng, Y.; Wu, S.; Shen, X.; He, J.; Luo, X. Hypoxia-related miR-210-5p and miR-210-3p regulate hypoxia-induced migration and epithelial-mesenchymal transition in hepatoma cells. Int. J. Clin. Exp. Med. 2019, 12, 5096–5104. [Google Scholar]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cao, Z.; Zhang, X.-M.; Nakamura, M.; Sun, M.; Hartman, J.; Harris, R.A.; Sun, Y.; Cao, Y. Novel mechanism of macrophage-mediated metastasis revealed in a zebrafish model of tumor development. Cancer Res. 2015, 75, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Dehari, H.; Sasaki, T.; Igarashi, T.; Ogi, K.; Okamoto, J.; Kawata, M.; Kobayashi, J.; Miyazaki, A.; Nakamori, K.; et al. Hypoxia-induced epithelial-mesenchymal transition is regulated by phosphorylation of GSK3-β via PI3 K/Akt signaling in oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, S.; Wang, Z.; Wang, F.; Cao, X.; Wu, Q.; Zhao, C.; Ma, H.; Ye, F.; Wang, H. Supervillin promotes epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma in hypoxia via activation of the RhoA/ROCK-ERK/p38 pathway. J. Exp. Clin. Cancer Res. 2018, 37, 128. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Cao, L.; Xiao, L.; Song, J.; Zhang, Y.; Zheng, P.; Zheng, S. Hypoxia induces actin cytoskeleton remodeling by regulating the binding of CAPZA1 to F-actin via PIP2 to drive EMT in hepatocellular carcinoma. Cancer Lett. 2019, 448, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gonciar, D.; Mocan, T.; Matea, C.T.; Zdrehus, C.; Mosteanu, O.; Mocan, L.; Pop, T. Nanotechnology in metastatic cancer treatment: Current Achievements and Future Research Trends. J. Cancer 2019, 10, 1358–1369. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365. [Google Scholar] [CrossRef] [Green Version]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sun, M.; Wang, L.; Jiao, B. HIFs, angiogenesis, and cancer. J. Cell. Biochem. 2013, 114, 967–974. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.-Q.; Woodard, A.S.; Fornaro, M.; Tallini, G.; Languino, L.R. Prostatic Carcinoma Cell Migration via αvβ3Integrin Is Modulated by a Focal Adhesion Kinase Pathway. Cancer Res. 1999, 59, 1655–1664. [Google Scholar]
- Befani, C.; Liakos, P. Hypoxia upregulates integrin gene expression in microvascular endothelial cells and promotes their migration and capillary-like tube formation. Cell Biol. Int. 2017, 41, 769–778. [Google Scholar] [CrossRef]
- Wan, J.; Chai, H.; Yu, Z.; Ge, W.; Kang, N.; Xia, W.; Che, Y. HIF-1α effects on angiogenic potential in human small cell lung carcinoma. J. Exp. Clin. Cancer Res. 2011, 30, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Sun, H.; Su, L.; Liu, L.; Shan, J.; Shen, J.; Yang, Z.; Chen, J.; Zhong, X.; Avila, M.A.; et al. Myocyte enhancer factor 2D promotes colorectal cancer angiogenesis downstream of hypoxia-inducible factor 1α. Cancer Lett. 2017, 400, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, Y.; Wada, H.; Eguchi, H.; Gotoh, K.; Kobayashi, S.; Kinoshita, M.; Kubo, M.; Hayashi, K.; Iwagami, Y.; Yamada, D.; et al. Exosomal miR-155 Derived from Hepatocellular Carcinoma Cells Under Hypoxia Promotes Angiogenesis in Endothelial Cells. Dig. Dis. Sci. 2019, 64, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, G.; Liu, S.; Su, H.; Wang, Y.; Li, J.; Luo, C. Remodeling the Tumor Microenvironment with Emerging Nanotherapeutics. Trends Pharm. Sci. 2018, 39, 59–74. [Google Scholar] [CrossRef]
- Pan, Y.; Wu, Q.; Qin, L.; Cai, J.; Du, B. Gold nanoparticles inhibit VEGF165-induced migration and tube formation of endothelial cells via the Akt pathway. BioMed Res. Int. 2014, 2014, 418624. [Google Scholar] [CrossRef] [Green Version]
- Darweesh, R.S.; Ayoub, N.M.; Nazzal, S. Gold nanoparticles and angiogenesis: Molecular mechanisms and biomedical applications. Int. J. Nanomed. 2019, 14, 7643–7663. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Rocha, L.A.; Learmonth, D.A.; Sousa, R.A.; Salgado, A.J. alphavbeta3 and alpha5beta1 integrin-specific ligands: From tumor angiogenesis inhibitors to vascularization promoters in regenerative medicine? Biotechnol. Adv. 2018, 36, 208–227. [Google Scholar] [CrossRef]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeltzing, O.; Liu, W.; Reinmuth, N.; Fan, F.; Parry, G.C.; Parikh, A.A.; McCarty, M.F.; Bucana, C.D.; Mazar, A.P.; Ellis, L.M. Inhibition of integrin α5β1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int. J. Cancer 2003, 104, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Umansky, V.; Kieda, C. The role of hypoxia in shaping the recruitment of proangiogenic and immunosuppressive cells in the tumor microenvironment. Contemp. Oncol. 2018, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 2016, 7, 75763. [Google Scholar] [CrossRef] [Green Version]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia enhances immunosuppression by inhibiting CD4+ effector T cell function and promoting Treg activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten pathways. Oncogene 2018, 37, 4239. [Google Scholar] [CrossRef]
- Zhu, G.; Tang, Y.; Geng, N.; Zheng, M.; Jiang, J.; Li, L.; Li, K.; Lei, Z.; Chen, W.; Fan, Y.; et al. HIF-α/MIF and NF-κB/IL-6 axes contribute to the recruitment of CD11b+ Gr-1+ myeloid cells in hypoxic microenvironment of HNSCC. Neoplasia 2014, 16, 168. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef] [Green Version]
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Muraille, E.; Leo, O.; Moser, M. TH1/TH2 paradigm extended: Macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front. Immunol. 2014, 5, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung-Cancer-derived extracellular vesicle MicroRNA-103a increases the oncogenic effects of macrophages by targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wu, S.; Yan, R.; Fan, L.; Yu, L.; Zhang, Y.; Wei, W.; Zhou, C.; Wu, X.; Zhong, M.; et al. The role of the hypoxia-Nrp-1 axis in the activation of M2-like tumor-associated macrophages in the tumor microenvironment of cervical cancer. Mol. Carcinog. 2019, 58, 388–397. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 1, 5158–5173. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Ruf, M.; Moch, H.; Schraml, P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int. J. Cancer 2016, 139, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Laakkonen, P.; Åkerman, M.E.; Biliran, H.; Yang, M.; Ferrer, F.; Karpanen, T.; Hoffman, R.M.; Ruoslahti, E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9381–9386. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Yu, X.; Jin, C.; Yang, F.; Fu, D.; Long, J.; Xu, J.; Zhan, C.; Lu, W. LyP-1-conjugated nanoparticles for targeting drug delivery to lymphatic metastatic tumors. Int. J. Pharm. 2010, 385, 150–156. [Google Scholar] [CrossRef]
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. αvβ3-integrin regulates PD-L1 expression and is involved in cancer immune evasion. Proc. Natl. Acad. Sci. USA 2019, 116, 20141–20150. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.-P.; et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat. Cell Biol. 2019, 21, 498. [Google Scholar] [CrossRef]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [Green Version]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.-T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Gonçalves, V.; Granja, S.; Martinho, O.; Honavar, M.; Pojo, M.; Costa, B.M.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget 2016, 7, 46335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Lee, I.; Bang, H.; Kim, H.C.; Lee, W.Y.; Yun, S.H.; Lee, J.; Lee, S.J.; Park, Y.S.; Kim, K.-M. MCT4 expression is a potential therapeutic target in colorectal cancer with peritoneal carcinomatosis. Mol. Cancer Ther. 2018, 17, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, Y.; Yamamoto, K.; Kogure, K.; Ichihara, J.; Terada, H. Steady state transcript levels of the type II hexokinase and type 1 glucose transporter in human tumor cell lines. Cancer Lett. 1994, 82, 27–32. [Google Scholar] [CrossRef]
- Fan, T.; Sun, G.; Sun, X.; Zhao, L.; Zhong, R.; Peng, Y. Tumor energy metabolism and potential of 3-Bromopyruvate as an inhibitor of aerobic glycolysis: Implications in tumor treatment. Cancers 2019, 11, 317. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P. Monocarboxylic acid transport. Compr. Physiol. 2011, 3, 1611–1643. [Google Scholar]
- Liu, Y.; Ji, X.; Tong, W.W.L.; Askhatova, D.; Yang, T.; Cheng, H.; Wang, Y.; Shi, J. Engineering multifunctional RNAi nanomedicine to concurrently target cancer hallmarks for combinatorial therapy. Angew. Chem. Int. Ed. 2018, 57, 1510–1513. [Google Scholar] [CrossRef]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, R.P.; Tatham, L.M.; Savage, A.C.; Tripathi, A.K.; Mlambo, G.; Ippolito, M.M.; Nenortas, E.; Rannard, S.P.; Owen, A.; Shapiro, T.A. Long-acting injectable atovaquone nanomedicines for malaria prophylaxis. Nat. Commun. 2018, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Calvo, J.; Lavandera, J.L.; Agüeros, M.; Irache, J.M. Cyclodextrin/poly (anhydride) nanoparticles as drug carriers for the oral delivery of atovaquone. Biomed. Microdevices 2011, 13, 1015–1025. [Google Scholar] [CrossRef]
- Zannella, V.E.; Dal Pra, A.; Muaddi, H.; McKee, T.D.; Stapleton, S.; Sykes, J.; Glicksman, R.; Chaib, S.; Zamiara, P.; Milosevic, M. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin. Cancer Res. 2013, 19, 6741–6750. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Xiao, Q.; Zheng, X.; Zhang, L.; Xing, H.; Ni, D.; Liu, Y.; Zhang, S.; Ren, Q.; Hua, Y.; et al. Single W18O49 nanowires: A multifunctional nanoplatform for computed tomography imaging and photothermal/photodynamic/radiation synergistic cancer therapy. Nano Res. 2015, 8, 3580–3590. [Google Scholar] [CrossRef]
- Si, J.; Shao, S.; Shen, Y.; Wang, K. Macrophages as active nanocarriers for targeted early and adjuvant cancer chemotherapy. Small 2016, 12, 5108–5119. [Google Scholar] [CrossRef]
- Overchuk, M.; Zheng, G. Overcoming obstacles in the tumor microenvironment: Recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials 2018, 156, 217–237. [Google Scholar] [CrossRef]
- Li, W.; Quan, Y.-Y.; Li, Y.; Lu, L.; Cui, M. Monitoring of tumor vascular normalization: The key points from basic research to clinical application. Cancer Manag. Res. 2018, 10, 4163. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Yuan, H.; Yang, Z.; von Roemeling, C.A.; Qie, Y.; Zhao, H.; Wang, Y.; Jiang, W.; Kim, B.Y.S. Therapeutic Remodeling of the Tumor Microenvironment Enhances Nanoparticle Delivery. Adv. Sci. 2019, 6, 1802070. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Hu, Y.; Pang, Z. Modulating the tumor microenvironment to enhance tumor nanomedicine delivery. Front. Pharmacol. 2017, 8, 952. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Nanoparticle Formulation | Drug/Therapeutic | Targeting Moiety | Target | Indications and Measured Benefit | Ref. |
|---|---|---|---|---|---|
| 2.1 Enrichment and propagation of cancer stem cells | |||||
| PLGA | Salinomycin | CD133 aptamer | CD133 receptor | Selectively kill CD133+ osteosarcoma cells et and in vivo and reduce tumoursphere formation and the percentage of Sao-2 CD133+ cells | [15] |
| PLGA-PEG | Salinomycin | CD133 Antibody | CD133 receptor | Reduction in the percentage of CD133+ ovarian cancer cells. 2.5- fold decrease in PA-1tumor sphere number compared to the saline control | [16] |
| Lipid polymers | Salinomycin | CD133 and EGFR aptamer | CD133 receptor EGFR | Targeting both osteosarcoma CSCs and cancer cells with high specificity, 90% decrease in tumour volume | [17] |
| Lipid polymers | ATRA | CD133 aptamer | CD133 receptor | Osteosarcoma tumour volume inhibitory rate for the ATRA-PLNP-CD133 treated group was 81.1% | [18] |
| Hyaluronic acid and styrene-maleic acid Nano micelle | Curcumin | Hyaluronic acid | CD44 receptor | Marked inhibition of NF-κB signalling and significant reduction in CD44+ expression cells in pancreatic cancer cells | [19] |
| Pluronic f127 | Doxorubicin | Chitosan | CD44 receptor | Increased the toxicity of doxorubicin (Dox)by six times compared to free Dox in eliminating CD44+ CSC-like cells in MCF-7 breast cancer (BCa) cells. | [20] |
| Liposome | Salinomycin Doxorubicin | A significant decrease in liver cancer stem cells in vivo (HepG2, HepG2-TS cells) | [21] | ||
| PLGA | Salinomycin Paclitaxel | Hyaluronic acid | CD44 receptor | A significant reduction in CD44+ cells in Breast cancer, MCF-7 and MDA-MB-231 cells | [22] |
| PEG-PLA | Doxorubicin ATRA | Induced differentiation of CSCs and sensitized cells toward DOX treatment. Combinatory treatment significantly reduces MDA-MB-231 tumour growth in vivo. | [23] | ||
| Mesoporous silica | Cisplatin, 5-fluoroucail, paclitaxel | siRNA | ABCG2 | Downregulation of ABCG2 significantly enhanced the drug-induced apoptosis and inhibited Hep-2 (laryngeal) tumour growth in vivo. | [24] |
| PLGA | Paclitaxel | Wedelolactone | SOX-2, ABCG2 | Wedelolactone treatment sensitizes MDA-MB-231 BCa cells to the effects of paclitaxel and significantly reduced the ALDH+ breast cancer CSCs and suppressed the tumour growth | [25] |
| Silica | γ-secretase inhibitor | Notch signalling | Breast cancer, MDA-MB-231 cells. Reduce ALDH side population in CAM model and suppressed tumor growth in vivo | [26] | |
| 2.2 Invasion and Metastasis | |||||
| AuNPs | MAPK signalling | Inhibited the proliferation of SKOV3 (ovarian) cancer cells and delayed the tumoral and metastases growth by reversing EMT and inhibition of MAPK signalling | [27] | ||
| PEI coated SPIONs | Src kinase, miR-21, MMP2 | Reduced the invadosome intensity and decreased the ability of Pan02 (pancreatic cancer) cells to invade through basement membrane. | [28] | ||
| FA-PEG-PEI-SPIONs | miR-125b-5p | JAK-STAT, Wnt/β-Catenin | Inhibited the invasion, migration, and growth of HCC HUH7 and HCCLM3 cells | [29] | |
| Zinc arsenite | SHP-1/JAK-STAT | Inhibit tumour growth of HCC xenografts by 2.2-fold and metastasis by 3.5-fold as compare free arsenic trioxide-based NPs | [30] | ||
| PEG-AuNPs | PI3/AKT | Supressed tumour growth and decrease sphere formation of glioblastoma and lung adenocarcinoma A549 cells | [31] | ||
| Hyaluronic acid conjugated NPs | cisplatin | siRNA | Snail, Twist | Knockdown Twist and reversed chemoresistance to reduce tumour growth and metastasis of Ovarian cancer, F2 and Ovacar 8 cells in vivo | [32] |
| Amphiphilic polymers | paclitaxel | siRNA | Snail, Twist | Inhibited tumour growth and metastasis of 4T1 tumours in vivo simultaneously | [33] |
| 2.3 Angiogenesis | |||||
| AuNPs | VEGF, bFGF | inhibited endothelial /fibroblast cell proliferation & angiogenesis in an ovarian cancer model in vivo | [34] | ||
| AuNPs | EMT, MMP-2 | Facilitated tumour vasculature normalization, increased blood perfusion and alleviate tumour hypoxia in a model of lung cancer (B16F10) in vivo | [35] | ||
| AuNPs | Anterior gradient 2 (AGR2) | Reduced vessel density, tumour volume and increased the pericyte coverage in metastatic CRC model (SW620) in vivo | [36] | ||
| AuNPs | RGD | αvβ3 | Induced tumour vascular disruption and improved the therapeutic outcome of radiotherapy of Panc-1 pancreatic tumours in vivo | [37] | |
| AuNPs | RGD | αvβ3 | Reduced breast cancer (MDA-MB-231) cell viability and increased DNA damage compared to radiation alone in vitro. | [38] | |
| 2.4 Immunosuppression | |||||
| β-cyclodextrin | TLR7/8 agonist (R848) | Cyclodextrin | Engulfed by TAMs | Remodelled TME from M2 to M1 phenotype. Improved anti-PD-1 response rates in murine colon cancer models | [39] |
| PLGA | TRL9 agonist | Galactose | MGL—TAMs | Reprogrammed TAMs from M2-M1, suppressed melanoma tumour growth and increased CTL infiltration in vivo | [40] |
| PLGA | PI3K-γ inhibitor (IPI-549) | AEAA | Sigma-1 receptor—TME | Reduced MDSC proportion and decreased tumour growth in pancreatic tumour model in vivo | [41] |
| Magnetic zinc-doped iron oxide | MDSC | Repolarise MDSCs from immunosuppressive to pro-inflammatory when combined with Rad. | [42] | ||
| PLGA | Tyrosine kinase inhibitor (Imatinib) | Lyp-1 | Nrp-1—Tregs | Enhanced tumour inhibition and survival of murine melanoma tumours when combined with immune checkpoint inhibitor | [43] |
| PLGA | Anti-PD-L1 & ICG | MMP-2 sensitive property | MMP-2—TME | Increased CTL tumour infiltration. Suppressed tumour growth and lung metastasis in 4T1 breast cancer model | [44] |
| 2.5 Metabolic reprogramming | |||||
| AuNPs | 3-BPP | HK2 (mitochondria) | Suppressed tumour cell glycolysis and OXPHOS in prostate cells in vitro | [45] | |
| PLGA | Atovaquone + Veterporfin | Complex III (mitochondria) | Improved intratumoural oxygenation and anti-tumour response to PDT in 4T1 tumour bearing mice | [46] | |
| Gelatin | Atovaquone + ICG | Complex III (mitochondria) | Improved intratumoural oxygenation and anti-tumour response to PDT in HeLa xenografts | [47] | |
| PEG-PCL | Metformin + IR780 | Complex I (mitochondria) | Decreased endogenous oxygen consumption in gastric cancer cells in vitro. Improved PDT and PTT in vivo | [48] | |
| Tungsten oxide (W18O49) | Metformin | Complex I (mitochondria) | Lowered OCR and inhibit tumour growth in Raji-lymphoma-bearing mice | [49] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, J.; Byrne, N.M.; Al Jamal, W.; Coulter, J.A. Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development. Cancers 2019, 11, 1989. https://doi.org/10.3390/cancers11121989
Feng J, Byrne NM, Al Jamal W, Coulter JA. Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development. Cancers. 2019; 11(12):1989. https://doi.org/10.3390/cancers11121989
Chicago/Turabian StyleFeng, Jie, Niall M. Byrne, Wafa Al Jamal, and Jonathan A. Coulter. 2019. "Exploiting Current Understanding of Hypoxia Mediated Tumour Progression for Nanotherapeutic Development" Cancers 11, no. 12: 1989. https://doi.org/10.3390/cancers11121989