Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications

 ,
,
Abstract
:1. Introduction
2. CSCs in Endometrial Cancer: Role and Biomarkers
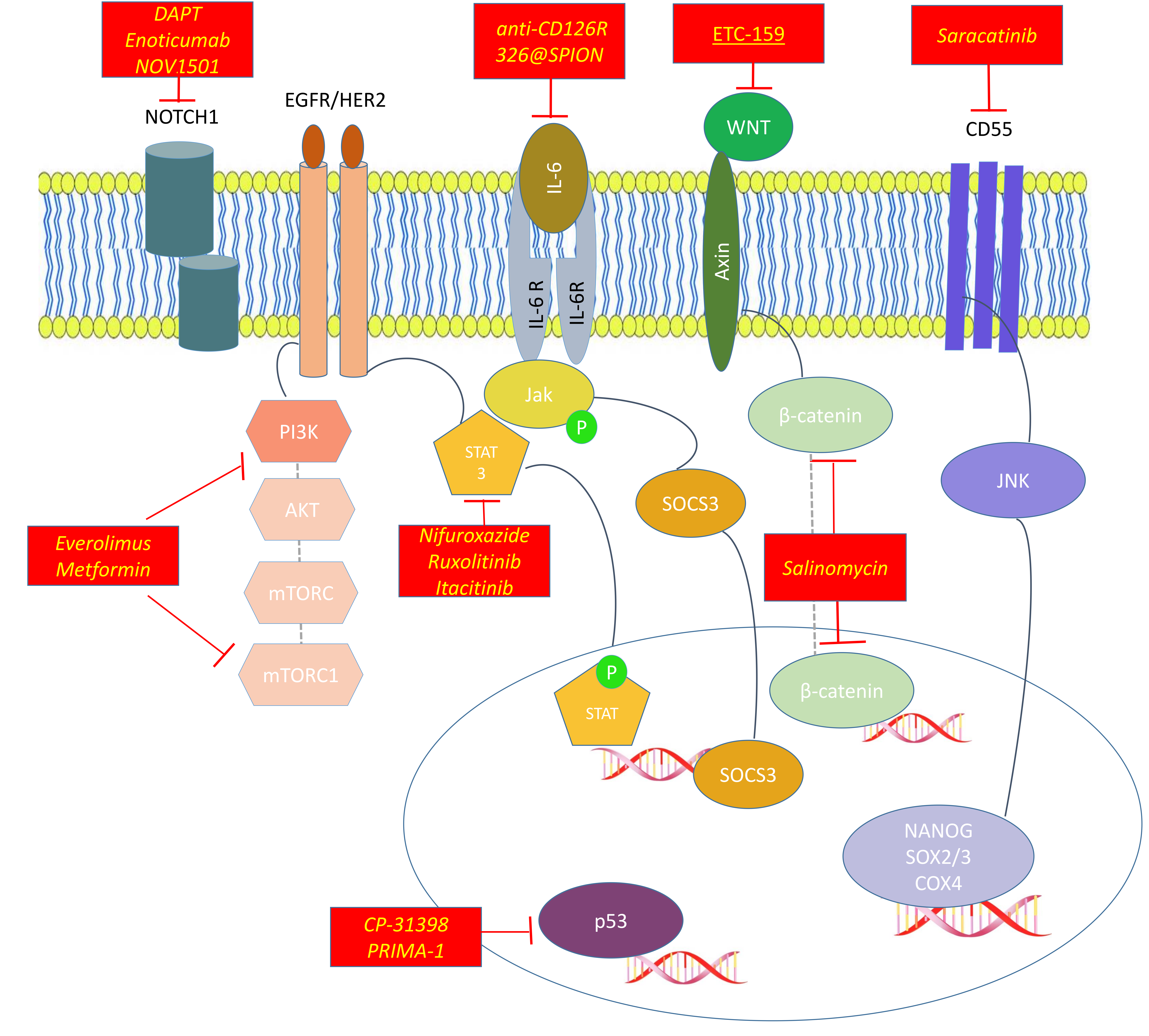
3. Activated Pathways in Endometrial CSCs
- Hedgehog signaling pathway, with its downstream effectors SMO and Gli1; its inhibitors are sonidegib and cyclopamine.
- PI3K/AKT/mTOR Complex (mTORC)pathway, inhibited by everolimus and metformin.
- Wnt/β-catenin pathway, inhibited by salinomycin and ETC-159.
- NOTCH pathway whose inhibitors are DAPT ((N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester))), Enoticumab and NOV1501 (also ALB001).
4. Agents Targeting CSCs in EC: Preclinical Development
5. Agents Targeting CSCs in EC: Clinical Trials and New Perspectives
6. Discussion
- Which is the best biomarker to detect CSCs?
- Is there a dependency on a specific pathway?
- Which is the most promising strategy to target CSCs?
- Probably CSCs heterogeneity justifies the presence of multiple biomarkers [85]. Indeed, CD133 and ALDH1 are the most studied and their expression correlates with prognosis and aggressiveness [63,83]. On the other hand markers of EMT [60] and expression of efflux pumps of ABCs family are certain markers of CSCs suggesting that the definition of a cluster of biomarkers could be the best strategy to detect CSCs in EC.
- CSCs related pathways are multiple and crosstalking, suggesting that there is no strict dependency on a specific pathway. Wnt-βcatenin [76], Notch [89] and Hedgehog [90] are the most important but also other signaling cascades implied in multiple biological mechanism like ER [113] and PI3K/AKT pathways [94] are involved in maintaining endometrial CSCs with a main role played also by epigenetic modulators.
- Nowadays we have few clinical data on drugs targeting CSCs. Certainly, most early phase studies showed that these drugs are safe but data on activity are lacking. Crosstalk and redundant pathways maintaining stemness are probably intrinsic mechanisms of resistance in several cancers and next therapeutic strategies could consider combination of drugs. The addition of drugs against CSC pathways to chemotherapy could be a tool to overcome resistance in ECs. An even more promising combination is the one of checkpoint inhibitors with drugs targeting CSCs above all in inflamed tumors among which MSI-high EC is one of the most interesting example [15,130]. Another powerful strategy could be treatment with chimeric antigen receptor-modified T (CART) cells, that recognize specific antigens, inducing T cell activation and survival, thus resulting in antitumor activity [44]. Preclinical works suggest that CSC-targeted CART cells against CD133 are effective in peritoneal carcinomatosis from gynecological cancers PDXs [131] but there is no ongoing study with CART133 including patients with ECs.
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cancer Stat Facts: Uterine Cancer. Available online: https://seer.cancer.gov/statfacts/html/corp.html (accessed on 28 September 2019).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, J.N.; Temkin, S.M.; Mackay, H.J. Endometrial cancer: Not your grandmother’s cancer. Cancer 2016, 122, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.M.; Overbeek-Wager, E.A.; Grumbo, R.J. Diagnosis and Management of Endometrial Cancer. Am. Fam. Physician 2016, 93, 468–474. [Google Scholar] [PubMed]
- Lu, K.H.; Schorge, J.O.; Rodabaugh, K.J.; Daniels, M.S.; Sun, C.C.; Soliman, P.T.; White, K.G.; Luthra, R.; Gershenson, D.M.; Broaddus, R.R. Prospective determination of prevalence of lynch syndrome in young women with endometrial cancer. J. Clin. Oncol. 2007, 25, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Signorelli, M.; Lissoni, A.A.; Cormio, G.; Katsaros, D.; Pellegrino, A.; Selvaggi, L.; Ghezzi, F.; Scambia, G.; Zola, P.; Grassi, R.; et al. Modified radical hysterectomy versus extrafascial hysterectomy in the treatment of stage I endometrial cancer: Results from the ILIADE randomized study. Ann. Surg. Oncol. 2009, 16, 3431–3441. [Google Scholar] [CrossRef]
- Nout, R.A.; Smit, V.T.; Putter, H.; Jürgenliemk-Schulz, I.M.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Mens, J.W.; Slot, A.; Kroese, M.C.; et al. Vaginal brachytherapy versus pelvic external beam radiotherapy for patients with endometrial cancer of high-intermediate risk (PORTEC-2): An open-label, non-inferiority, randomised trial. Lancet 2010, 375, 816–823. [Google Scholar] [CrossRef]
- De Boer, S.M.; Powell, M.E.; Mileshkin, L.; Katsaros, D.; Bessette, P.; Haie-Meder, C.; Ottevanger, P.B.; Ledermann, J.A.; Khaw, P.; Colombo, A.; et al. Adjuvant chemoradiotherapy versus radiotherapy alone for women with high-risk endometrial cancer (PORTEC-3): Final results of an international, open-label, multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 295–309. [Google Scholar] [CrossRef]
- National Comperhensive Cancer Network. Available online: https://www.nccn.org/professionals/physician_gls/pdf/uterine_blocks.pdf (accessed on 28 September 2019).
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef]
- Fung-Kee-Fung, M.; Dodge, J.; Elit, L.; Lukka, H.; Chambers, A.; Oliver, T.; Cancer Care Ontario Program in Evidence-based Care Gynecology Cancer Disease Site Group. Follow-up after primary therapy for endometrial cancer: A systematic review. Gynecol. Oncol. 2006, 101, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Lajer, H.; Jensen, M.B.; Kilsmark, J.; Albæk, J.; Svane, D.; Mirza, M.R.; Geertsen, P.F.; Reerman, D.; Hansen, K.; Milter, M.C.; et al. The value of gynecologic cancer follow-up: Evidence-based ignorance? Int. J. Gynecol. Cancer 2010, 20, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Aglietta, M.; Genta, S.; Valabrega, G. Checkpoint inhibitors in endometrial cancer: Preclinical rationale and clinical activity. Oncotarget 2017, 8, 90532–90544. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, P.; Della Pepa, C.; Berardi, S.; Califano, D.; Scala, S.; Buonaguro, L.; Ciliberto, G.; Brauchli, P.; Pignata, S. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat. Rev. 2016, 48, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Keytruda. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125514s065lbl.pdf (accessed on 14 November 2019).
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Wang, T.; Shigdar, S.; Gantier, M.P.; Hou, Y.; Wang, L.; Li, Y.; Shamaileh, H.A.; Yin, W.; Zhou, S.F.; Zhao, X.; et al. Cancer stem cell targeted therapy: Progress amid controversies. Oncotarget 2015, 6, 44191–44206. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ito, T.; Zimdahl, B.; Reya, T. aSIRTing control over cancer stem cells. Cancer Cell 2012, 21, 140–142. [Google Scholar] [CrossRef]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncol. Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Hu, J.; Sun, F.; Chen, W.; Zhang, J.; Zhang, T.; Qi, M.; Feng, T.; Liu, H.; Li, X.; Xing, Y.; et al. BTF3 sustains cancer stem-like phenotype of prostate cancer via stabilization of BMI1. J. Exp. Clin. Cancer Res. 2019, 38, 227. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Marzagalli, M.; Raimondi, M.; Fontana, F.; Montagnani Marelli, M.; Moretti, R.M.; Limonta, P. Cellular and molecular biology of cancer stem cells in melanoma: Possible therapeutic implications. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Brown, H.K.; Tellez-Gabriel, M.; Heymann, D. Cancer stem cells in osteosarcoma. Cancer Lett. 2017, 386, 189–195. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.L.; Ouyang, R.R.; Chen, S.J.; Gu, Y.Z.; Huang, L.A.; Lu, J.X.; Wang, Z.Y. Treatment of acute promyelocytic leukemia with all-trans retinoic acid. A five-year experience. Chin. Med. J. (Engl.) 1993, 106, 743–748. [Google Scholar] [PubMed]
- Huang, F.; Zhao, H.P.; Gao, X.Z.; Dai, M.M.; Fan, L.L. Recombinant human G-CSF and retinoic acid in synergistically inducing granulocyte differentiation of human promyelocytic leukemic cells. Chin. Med. J. (Engl.) 1992, 105, 707–712. [Google Scholar]
- Reichardt, P. The Story of Imatinib in GIST—A Journey through the Development of a Targeted Therapy. Oncol. Res. Treat. 2018, 41, 472–477. [Google Scholar] [CrossRef]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in Cancer: A Hematological Perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015, 6, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Feng, K.; Wang, Y.; Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: A potential and curable approach for cancer treatment. Protein Cell 2018, 9, 516–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zheng, H.; Luo, W.; Zhang, Q.; Liu, J.; Yao, K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci. Rep. 2017, 7, 4859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapdor, R.; Wang, S.; Hacker, U.; Büning, H.; Morgan, M.; Dörk, T.; Hillemanns, P.; Schambach, A. Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy. Hum. Gene Ther. 2017, 28, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.A.; Friel, A.M.; Kumar, B.; Zhang, L.; Rueda, B.R.; Gargett, C.E. Evidence for cancer stem cells in human endometrial carcinoma. Cancer Res. 2009, 69, 8241–8248. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, M.J.; Laranjo, M.; Abrantes, A.M.; Torgal, I.; Botelho, M.F.; Oliveira, C.F. Clinical translation for endometrial cancer stem cells hypothesis. Cancer Metastasis Rev. 2015, 34, 401–416. [Google Scholar] [CrossRef]
- Hubbard, S.A.; Gargett, C.E. A cancer stem cell origin for human endometrial carcinoma? Reproduction 2010, 140, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Allegra, A.; Alonci, A.; Penna, G.; Innao, V.; Gerace, D.; Rotondo, F.; Musolino, C. The cancer stem cell hypothesis: A guide to potential molecular targets. Cancer Investig. 2014, 32, 470–495. [Google Scholar] [CrossRef] [PubMed]
- Tempest, N.; Maclean, A.; Hapangama, D.K. Endometrial Stem Cell Markers: Current Concepts and Unresolved Questions. Int. J. Mol. Sci. 2018, 19, 3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edling, C.E.; Hallberg, B. c-Kit--a hematopoietic cell essential receptor tyrosine kinase. Int. J. Biochem. Cell Biol. 2007, 39, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Levina, V.; Marrangoni, A.; Wang, T.; Parikh, S.; Su, Y.; Herberman, R.; Lokshin, A.; Gorelik, E. Elimination of human lung cancer stem cells through targeting of the stem cell factor-c-kit autocrine signaling loop. Cancer Res. 2010, 70, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kyo, S.; Nakamura, M.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Fujiwara, H. Imatinib sensitizes endometrial cancer cells to cisplatin by targeting CD117-positive growth-competent cells. Cancer Lett. 2014, 345, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Mirantes, C.; Espinosa, I.; Ferrer, I.; Dolcet, X.; Prat, J.; Matias-Guiu, X. Epithelial-to-mesenchymal transition and stem cells in endometrial cancer. Hum. Pathol. 2013, 44, 1973–1981. [Google Scholar] [CrossRef]
- Park, J.Y.; Hong, D. Association between Morphological Patterns of Myometrial Invasion and Cancer Stem Cell Markers in Endometrial Endometrioid Carcinoma. Pathol. Oncol. Res. 2019, 25, 123–130. [Google Scholar] [CrossRef]
- Elbasateeny, S.S.; Salem, A.A.; Abdelsalam, W.A.; Salem, R.A. Immunohistochemical expression of cancer stem cell related markers CD44 and CD133 in endometrial cancer. Pathol. Res. Pr. 2016, 212, 10–16. [Google Scholar] [CrossRef]
- Rutella, S.; Bonanno, G.; Procoli, A.; Mariotti, A.; Corallo, M.; Prisco, M.G.; Eramo, A.; Napoletano, C.; Gallo, D.; Perillo, A.; et al. Cells with characteristics of cancer stem/progenitor cells express the CD133 antigen in human endometrial tumors. Clin. Cancer Res. 2009, 15, 4299–4311. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Wiechert, A.; Rao, V.S.; Alluri, R.; Connor, E.; Thiagarajan, P.S.; Hale, J.S.; Li, Y.; Chumakova, A.; Jarrar, A.; et al. CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors. J. Exp. Med. 2017, 214, 2715–2732. [Google Scholar] [CrossRef] [PubMed]
- Mizrak, D.; Brittan, M.; Alison, M. CD133: Molecule of the moment. J. Pathol. 2008, 214, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Martinelli, E.; Petrillo, M.; Prisco, M.G.; Zannoni, G.; Sioletic, S.; Scambia, G. CD133 antigen expression in ovarian cancer. BMC Cancer 2009, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Friel, A.M.; Zhang, L.; Curley, M.D.; Therrien, V.A.; Sergent, P.A.; Belden, S.E.; Borger, D.R.; Mohapatra, G.; Zukerberg, L.R.; Foster, R.; et al. Epigenetic regulation of CD133 and tumorigenicity of CD133 positive and negative endometrial cancer cells. Reprod. Biol. Endocrinol. 2010, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Kyo, S.; Zhang, B.; Zhang, X.; Mizumoto, Y.; Takakura, M.; Maida, Y.; Mori, N.; Hashimoto, M.; Ohno, S.; et al. Prognostic impact of CD133 expression as a tumor-initiating cell marker in endometrial cancer. Hum. Pathol. 2010, 41, 1516–1529. [Google Scholar] [CrossRef]
- Kyo, S.; Maida, Y.; Inoue, M. Stem cells in endometrium and endometrial cancer: Accumulating evidence and unresolved questions. Cancer Lett. 2011, 308, 123–133. [Google Scholar] [CrossRef]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Turnquist, H.; Jackson, J.; Sgagias, M.; Yan, Y.; Gong, M.; Dean, M.; Sharp, J.G.; Cowan, K. The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin. Cancer Res. 2002, 8, 22–28. [Google Scholar]
- Doyle, L.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Takao, T.; Kuboyama, A.; Tanaka, Y.; Ohgami, T.; Yamaguchi, S.; Adachi, S.; Yoneda, T.; Ueoka, Y.; Hayashi, S.; et al. Endometrial cancer side-population cells show prominent migration and have a potential to differentiate into the mesenchymal cell lineage. Am. J. Pathol. 2010, 176, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusunoki, S.; Kato, K.; Tabu, K.; Inagaki, T.; Okabe, H.; Kaneda, H.; Suga, S.; Terao, Y.; Taga, T.; Takeda, S. The inhibitory effect of salinomycin on the proliferation, migration and invasion of human endometrial cancer stem-like cells. Gynecol. Oncol. 2013, 129, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Zhang, X.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Kyo, S. Molecular characterization of CD133+ cancer stem-like cells in endometrial cancer. Int. J. Oncol. 2014, 44, 669–677. [Google Scholar] [CrossRef]
- Yusuf, N.; Inagaki, T.; Kusunoki, S.; Okabe, H.; Yamada, I.; Matsumoto, A.; Terao, Y.; Takeda, S.; Kato, K. SPARC was overexpressed in human endometrial cancer stem-like cells and promoted migration activity. Gynecol. Oncol. 2014, 134, 356–363. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ginestier, C.; Charafe-Jauffret, E.; Foco, H.; Kleer, C.G.; Merajver, S.D.; Dontu, G.; Wicha, M.S. BRCA1 regulates human mammary stem/progenitor cell fate. Proc. Natl. Acad. Sci. USA 2008, 105, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Rahadiani, N.; Ikeda, J.; Mamat, S.; Matsuzaki, S.; Ueda, Y.; Umehara, R.; Tian, T.; Wang, Y.; Enomoto, T.; Kimura, T.; et al. Expression of aldehyde dehydrogenase 1 (ALDH1) in endometrioid adenocarcinoma and its clinical implications. Cancer Sci. 2011, 102, 903–908. [Google Scholar] [CrossRef]
- Kitson, S.J.; Rosser, M.; Fischer, D.P.; Marshall, K.M.; Clarke, R.B.; Crosbie, E.J. Targeting Endometrial Cancer Stem Cell Activity with Metformin Is Inhibited by Patient-Derived Adipocyte-Secreted Factors. Cancers (Basel) 2019, 11, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, Y.; Hirohashi, Y.; Hashimoto, S.; Mariya, T.; Asano, T.; Ikeo, K.; Kuroda, T.; Mizuuchi, M.; Murai, A.; Uno, S.; et al. Clonal analysis revealed functional heterogeneity in cancer stem-like cell phenotypes in uterine endometrioid adenocarcinoma. Exp. Mol. Pathol. 2019, 106, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.F.; Li, D.; Yang, H.; Ma, J.; Pan, X.; Liu, H.X.; Huo, J.N.; Ma, X.X. Preliminary identification of endometrial cancer stem cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Coleman, R.L.; Harris, A.L.; Sood, A.K. Molecular pathways: Translational and therapeutic implications of the Notch signaling pathway in cancer. Clin. Cancer Res. 2015, 21, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götte, M.; Greve, B.; Kelsch, R.; Müller-Uthoff, H.; Weiss, K.; Kharabi Masouleh, B.; Sibrowski, W.; Kiesel, L.; Buchweitz, O. The adult stem cell marker Musashi-1 modulates endometrial carcinoma cell cycle progression and apoptosis via Notch-1 and p21WAF1/CIP1. Int. J. Cancer 2011, 129, 2042–2049. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, Y.P.; Huang, G.R.; Gong, B.L.; Yang, B.; Zhang, D.X.; Hu, P.; Xu, S.R. Expression of the stem cell marker, Nanog, in human endometrial adenocarcinoma. Int. J. Gynecol. Pathol. 2011, 30, 262–270. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef]
- Lu, H.; Ju, D.D.; Yang, G.D.; Zhu, L.Y.; Yang, X.M.; Li, J.; Song, W.W.; Wang, J.H.; Zhang, C.C.; Zhang, Z.G.; et al. Targeting cancer stem cell signature gene SMOC-2 Overcomes chemoresistance and inhibits cell proliferation of endometrial carcinoma. EBioMedicine 2019, 40, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Konno, Y.; Watari, H.; Hosaka, M.; Noguchi, M.; Sakuragi, N. The impact of microRNA-mediated PI3K/AKT signaling on epithelial-mesenchymal transition and cancer stemness in endometrial cancer. J. Transl. Med. 2014, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.Z.; Shiozawa, T.; Miyamoto, T.; Kashima, H.; Kurai, M.; Suzuki, A.; Ying-Song, J.; Konishi, I. Overexpression of hedgehog signaling molecules and its involvement in the proliferation of endometrial carcinoma cells. Clin. Cancer Res. 2007, 13, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Yan, L.; Zhao, X.; Li, C.; Fu, Y. microRNA-21 overexpression contributes to cell proliferation by targeting PTEN in endometrioid endometrial cancer. Oncol. Lett. 2012, 4, 1290–1296. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, M.J.; Laranjo, M.; Abrantes, A.M.; Casalta-Lopes, J.; Sarmento-Santos, D.; Costa, T.; Serambeque, B.; Almeida, N.; Gonçalves, T.; Mamede, C.; et al. Endometrial Cancer Spheres Show Cancer Stem Cells Phenotype and Preference for Oxidative Metabolism. Pathol. Oncol. Res. 2019, 25, 1163–1174. [Google Scholar] [CrossRef]
- Shiba, S.; Ikeda, K.; Suzuki, T.; Shintani, D.; Okamoto, K.; Horie-Inoue, K.; Hasegawa, K.; Inoue, S. Hormonal regulation of patient-derived endometrial cancer stem-like cells generated by three-dimensional culture. Endocrinology 2019. [Google Scholar] [CrossRef]
- Kiyohara, M.H.; Dillard, C.; Tsui, J.; Kim, S.R.; Lu, J.; Sachdev, D.; Goodglick, L.; Tong, M.; Torous, V.F.; Aryasomayajula, C.; et al. EMP2 is a novel therapeutic target for endometrial cancer stem cells. Oncogene 2017, 36, 5793–5807. [Google Scholar] [CrossRef] [Green Version]
- Ran, X.; Zhou, P.; Zhang, K. Autophagy plays an important role in stemness mediation and the novel dual function of EIG121 in both autophagy and stemness regulation of endometrial carcinoma JEC cells. Int. J. Oncol. 2017, 51, 644–656. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, T.; Huang, Y. MicroRNA-134 suppresses endometrial cancer stem cells by targeting POGLUT1 and Notch pathway proteins. FEBS Lett. 2015, 589, 207–214. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Huang, K.; Wang, Y.; Li, J.; Yang, X. MicroRNA-34a inhibits cells proliferation and invasion by downregulating Notch1 in endometrial cancer. Oncotarget 2017, 8, 111258–111270. [Google Scholar] [CrossRef]
- Shang, C.; Lang, B.; Meng, L.R. Blocking NOTCH pathway can enhance the effect of EGFR inhibitor through targeting CD133+ endometrial cancer cells. Cancer Biol. Ther. 2018, 19, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Hu, J.; Yu, T.; You, S.; Zhang, Y.; Hu, L. miR-27b-3p/MARCH7 regulates invasion and metastasis of endometrial cancer cells through Snail-mediated pathway. Acta Biochim. Biophys. Sin. 2019, 51, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Kato, K. Endometrial Cancer Stem Cell as a Potential Therapeutic Target. Semin. Reprod. Med. 2015, 33, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T.; Onishi, T. Estrogen induces cell proliferation by promoting ABCG2-mediated efflux in endometrial cancer cells. Biochem. Biophys. Rep. 2018, 16, 74–78. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Park, W.H.; Suh, D.H.; Kim, K.; Kim, Y.B.; No, J.H. Silencing Bmi1 expression suppresses cancer stemness and enhances chemosensitivity in endometrial cancer cells. Biomed. Pharm. 2018, 108, 584–589. [Google Scholar] [CrossRef]
- Buechel, M.; Dey, A.; Dwivedi, S.K.D.; Crim, A.; Ding, K.; Zhang, R.; Mukherjee, P.; Moore, K.N.; Cao, L.; Branstrom, A.; et al. Inhibition of BMI1, a Therapeutic Approach in Endometrial Cancer. Mol. Cancer Ther. 2018, 17, 2136–2143. [Google Scholar] [CrossRef] [Green Version]
- Van der Zee, M.; Sacchetti, A.; Cansoy, M.; Joosten, R.; Teeuwssen, M.; Heijmans-Antonissen, C.; Ewing-Graham, P.C.; Burger, C.W.; Blok, L.J.; Fodde, R. IL6/JAK1/STAT3 Signaling Blockade in Endometrial Cancer Affects the ALDHhi/CD126+ Stem-like Component and Reduces Tumor Burden. Cancer Res. 2015, 75, 3608–3622. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Qian, H.; Tang, X.; Du, X.; Wang, G.; Zhang, H.; Ye, F.; Liu, T. Superparamagnetic iron oxide nanoparticle-mediated expression of. Int. J. Nanomed. 2019, 14, 2719–2731. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Yue, Y.; Wang, R.; Zhang, Y.; Jin, Q.; Zhou, X. Overexpression of microRNA-194 suppresses the epithelial-mesenchymal transition in targeting stem cell transcription factor Sox3 in endometrial carcinoma stem cells. Tumor Biol. 2017, 39, 1010428317706217. [Google Scholar] [CrossRef] [Green Version]
- Guy, M.S.; Qamar, L.; Behbakht, K.; Post, M.D.; Sheeder, J.; Sartorius, C.A.; Spillman, M.A. Progestin treatment decreases CD133+ cancer stem cell populations in endometrial cancer. Gynecol. Oncol. 2016, 140, 518–526. [Google Scholar] [CrossRef]
- Kato, K.; Kuhara, A.; Yoneda, T.; Inoue, T.; Takao, T.; Ohgami, T.; Dan, L.; Kuboyama, A.; Kusunoki, S.; Takeda, S.; et al. Sodium butyrate inhibits the self-renewal capacity of endometrial tumor side-population cells by inducing a DNA damage response. Mol. Cancer Ther. 2011, 10, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Elit, L.; Tsao, M.S.; Kamel-Reid, S.; Biagi, J.; Provencher, D.M.; Gotlieb, W.H.; Hoskins, P.J.; Ghatage, P.; Tonkin, K.S.; et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. J. Clin. Oncol. 2011, 29, 3278–3285. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Favier, L.; Weber, B.; Roemer-Becuwe, C.; Bougnoux, P.; Fabbro, M.; Floquet, A.; Joly, F.; Plantade, A.; Paraiso, D.; et al. Everolimus as second- or third-line treatment of advanced endometrial cancer: ENDORAD, a phase II trial of GINECO. Br. J. Cancer 2013, 108, 1771–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, N.; McMeekin, D.S.; Schwartz, P.E.; Sessa, C.; Gehrig, P.A.; Holloway, R.; Braly, P.; Matei, D.; Morosky, A.; Dodion, P.F.; et al. Ridaforolimus as a single agent in advanced endometrial cancer: Results of a single-arm, phase 2 trial. Br. J. Cancer 2013, 108, 1021–1026. [Google Scholar] [CrossRef]
- Tsoref, D.; Welch, S.; Lau, S.; Biagi, J.; Tonkin, K.; Martin, L.A.; Ellard, S.; Ghatage, P.; Elit, L.; Mackay, H.J.; et al. Phase II study of oral ridaforolimus in women with recurrent or metastatic endometrial cancer. Gynecol. Oncol. 2014, 135, 184–189. [Google Scholar] [CrossRef]
- Fleming, G.F.; Filiaci, V.L.; Marzullo, B.; Zaino, R.J.; Davidson, S.A.; Pearl, M.; Makker, V.; Burke, J.J.; Zweizig, S.L.; Van Le, L.; et al. Temsirolimus with or without megestrol acetate and tamoxifen for endometrial cancer: A gynecologic oncology group study. Gynecol. Oncol. 2014, 132, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Slomovitz, B.M.; Jiang, Y.; Yates, M.S.; Soliman, P.T.; Johnston, T.; Nowakowski, M.; Levenback, C.; Zhang, Q.; Ring, K.; Munsell, M.F.; et al. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J. Clin. Oncol. 2015, 33, 930–936. [Google Scholar] [CrossRef]
- Meireles, C.G.; Pereira, S.A.; Valadares, L.P.; Rêgo, D.F.; Simeoni, L.A.; Guerra, E.N.; Lofrano-Porto, A. Effects of metformin on endometrial cancer: Systematic review and meta-analysis. Gynecol. Oncol. 2017, 147, 167–180. [Google Scholar] [CrossRef]
- Pamela, T.S.; Shannon Neville, W.; David, A.I.; Mark, F.M.; Brian, M.S.; Karen, H.L.; Robert, L.C. Phase II study of everolimus, letrozole, and metformin in women with advanced/recurrent endometrial cancer. J. Clin. Oncol. 2016, 34, 5506. [Google Scholar]
- Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 2353. [Google Scholar] [CrossRef] [Green Version]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slosberg, E.D.; Kang, B.P.; Peguero, J.; Taylor, M.; Bauer, T.M.; Berry, D.A.; Braiteh, F.; Spira, A.; Meric-Bernstam, F.; Stein, S.; et al. Signature program: A platform of basket trials. Oncotarget 2018, 9, 21383–21395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annex I Summary of Product Characteristic. Available online: https://www.ema.europa.eu/en/documents/product-information/odomzo-epar-product-information_en.pdf (accessed on 28 September 2019).
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Ng, M.; Subbiah, V.; Messersmith, W.; Teneggi, V. Extension study of ETC-159 an oral PORCN inhibitor administered with bone protective treatment, in patients with advanced solid tumours. Ann. Oncol. 2018, 29, mdy430-002.2018. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Codd, A.S.; Kanaseki, T.; Torigo, T.; Tabi, Z. Cancer stem cells as targets for immunotherapy. Immunology 2018, 153, 304–314. [Google Scholar] [CrossRef]
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.H.; Chen, C.; Tay, J.C.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget 2017, 8, 13545–13559. [Google Scholar] [CrossRef]


{kind=link}
| Description | Condition | Line of Therapy | Primary Endpoint | Phase | Status | Trial Identifier |
|---|---|---|---|---|---|---|
| Targeted Therapy Directed by Genetic Testing (Pi3k inhibitors, AKT inhibitors, Hedgehog Antagonist) | Advanced solid tumors | ≥2 line | ORR | II | Recruiting | NCT02465060 |
| DKN-01 +/− Paclitaxel | EC, OC or Carcinosarcoma | ≥2 line | ORR, toxicity, pharmacokinetic | II | Recruiting | NCT03395080 |
| ETC-159 +/− Pembrolizumab | EC, OC or colorectal cancer | ≥2 line | % AE | I | Active not recruiting | NCT02521844 |
| COTI-2 (Against p53) +/− cisplatin | EC or Other solid tumors | na | DLT, MTD, RP2D | I | Recruiting | NCT02433626 |
| Everolimus, Letrozole, and Metformin | EC | ≤3 line | CBR | II | Active not recruiting | NCT01797523 |
| Ribociclib+ Everolimus + Letrozole | EC | ≤ 3 line | DLT/CBR | I/II | Recruiting | NCT03008408 |
| Gedatolisib + Palbociclib | Solid tumors | NA | MTD, RP2D, %AE | I | Recruiting | NCT03065062 |
| Cyclophosphamide + Metformin + Olaparib | EC | ≥2 line | RP2D | I/II | Notyetrecruiting | NCT02755844 |
| AZD2014(MTOR inhibitor) + anastrozole | ER+ EC | ≥2 line | % AE, 8weeks PFS | I/II | Recruiting | NCT02730923 |
| NOV1501 (anti DLL-4) | Solid tumors | NA | DLT | I | Recruiting | NCT03292783 |
| Itacitinib (anti Jak 3) (INCB039110) and/or Pembrolizumab | Solid tumors | NA | Toxicity | I | Active not recruiting | NCT02646748 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannone, G.; Attademo, L.; Scotto, G.; Genta, S.; Ghisoni, E.; Tuninetti, V.; Aglietta, M.; Pignata, S.; Valabrega, G. Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications. Cancers 2019, 11, 1820. https://doi.org/10.3390/cancers11111820
Giannone G, Attademo L, Scotto G, Genta S, Ghisoni E, Tuninetti V, Aglietta M, Pignata S, Valabrega G. Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications. Cancers. 2019; 11(11):1820. https://doi.org/10.3390/cancers11111820
Chicago/Turabian StyleGiannone, Gaia, Laura Attademo, Giulia Scotto, Sofia Genta, Eleonora Ghisoni, Valentina Tuninetti, Massimo Aglietta, Sandro Pignata, and Giorgio Valabrega. 2019. "Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications" Cancers 11, no. 11: 1820. https://doi.org/10.3390/cancers11111820