WWOX Possesses N-Terminal Cell Surface-Exposed Epitopes WWOX7-21 and WWOX7-11 for Signaling Cancer Growth Suppression and Prevention In Vivo

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
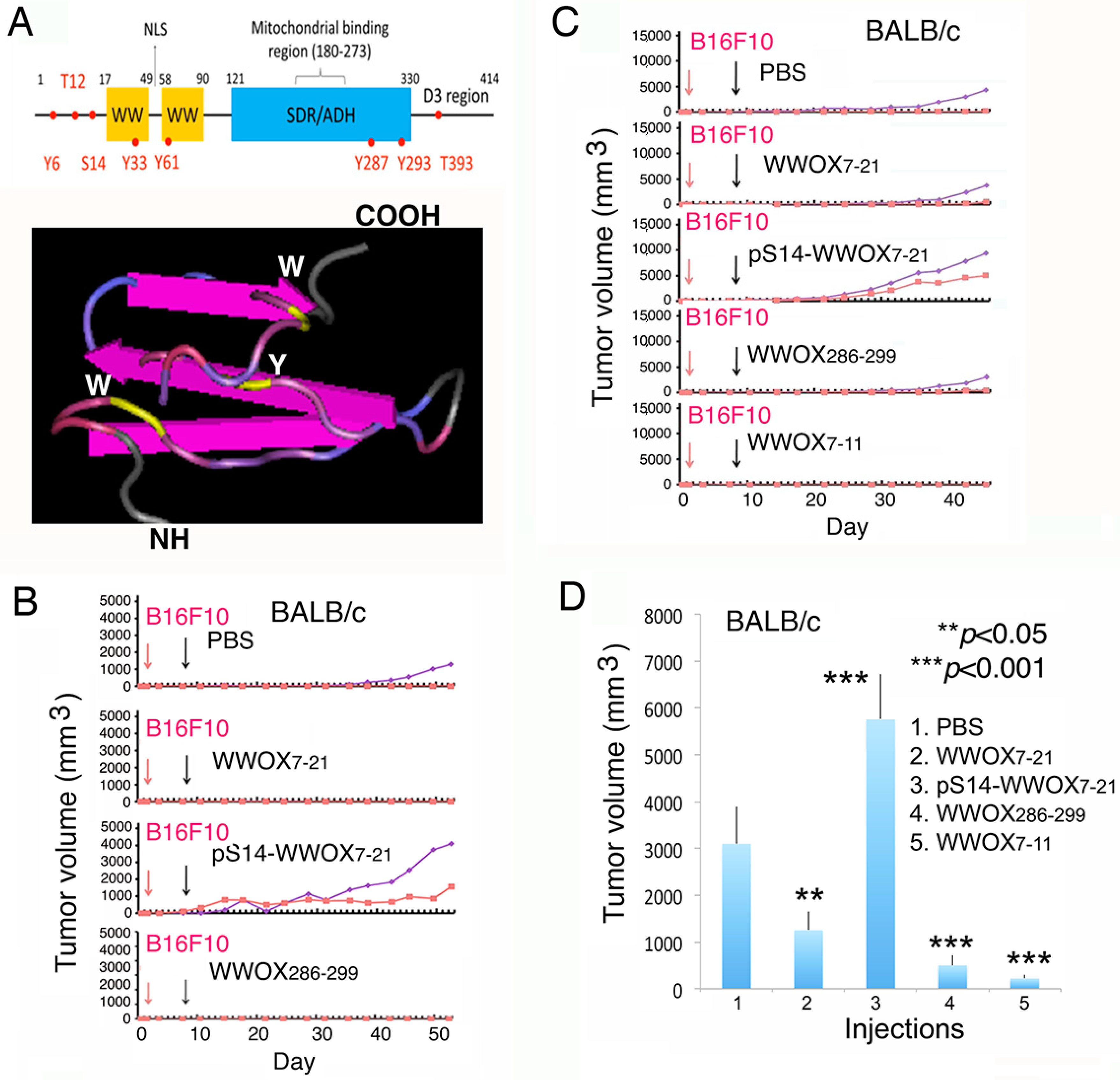
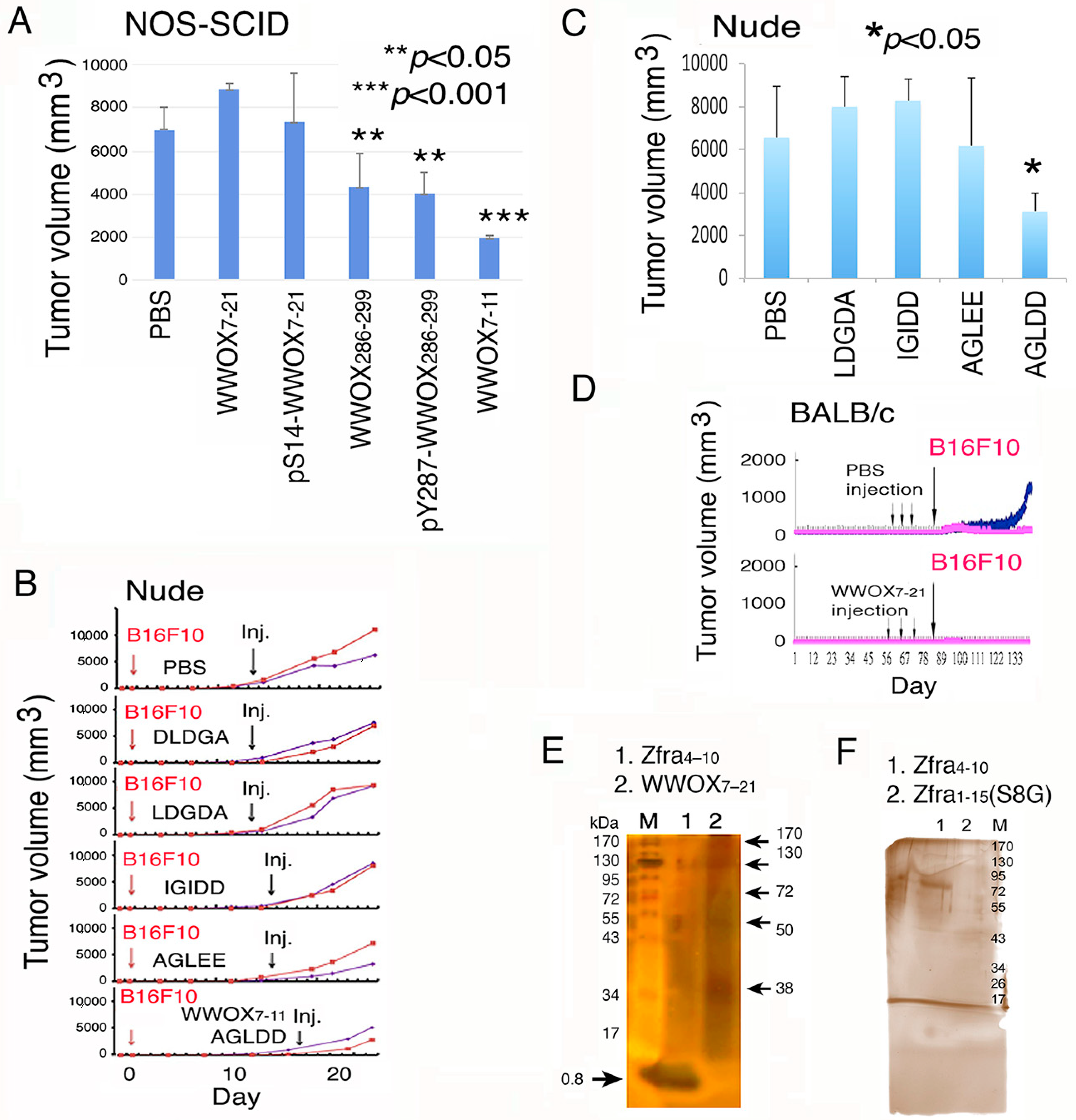
2.1. WWOX7-21 and WWOX7-11 Peptides Effectively Inhibit Melanoma Cell Growth in Both Immune Competent and Deficient Mice
2.2. WWOX7-21 Peptide Effectively Prevents Skin Cancer and Melanoma Cell Growth In Vivo
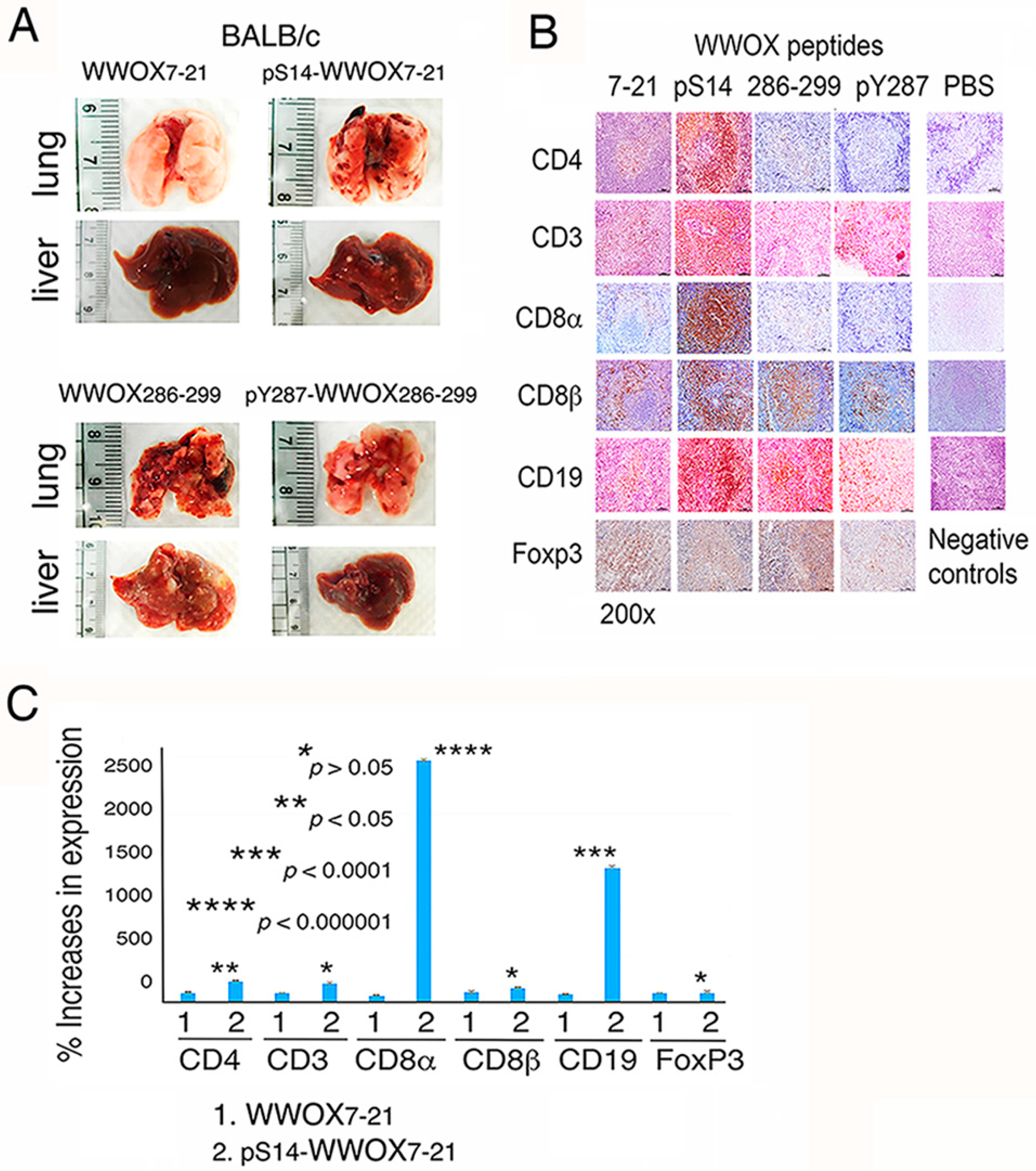
2.3. WWOX7-21 Peptide Blocks Melanoma Cell Metastasis
2.4. pS14-WWOX7-21 Peptide Dramatically Induces Cytotoxic T Cell Expansion but Fails to Block Cancer Cell Metastasis
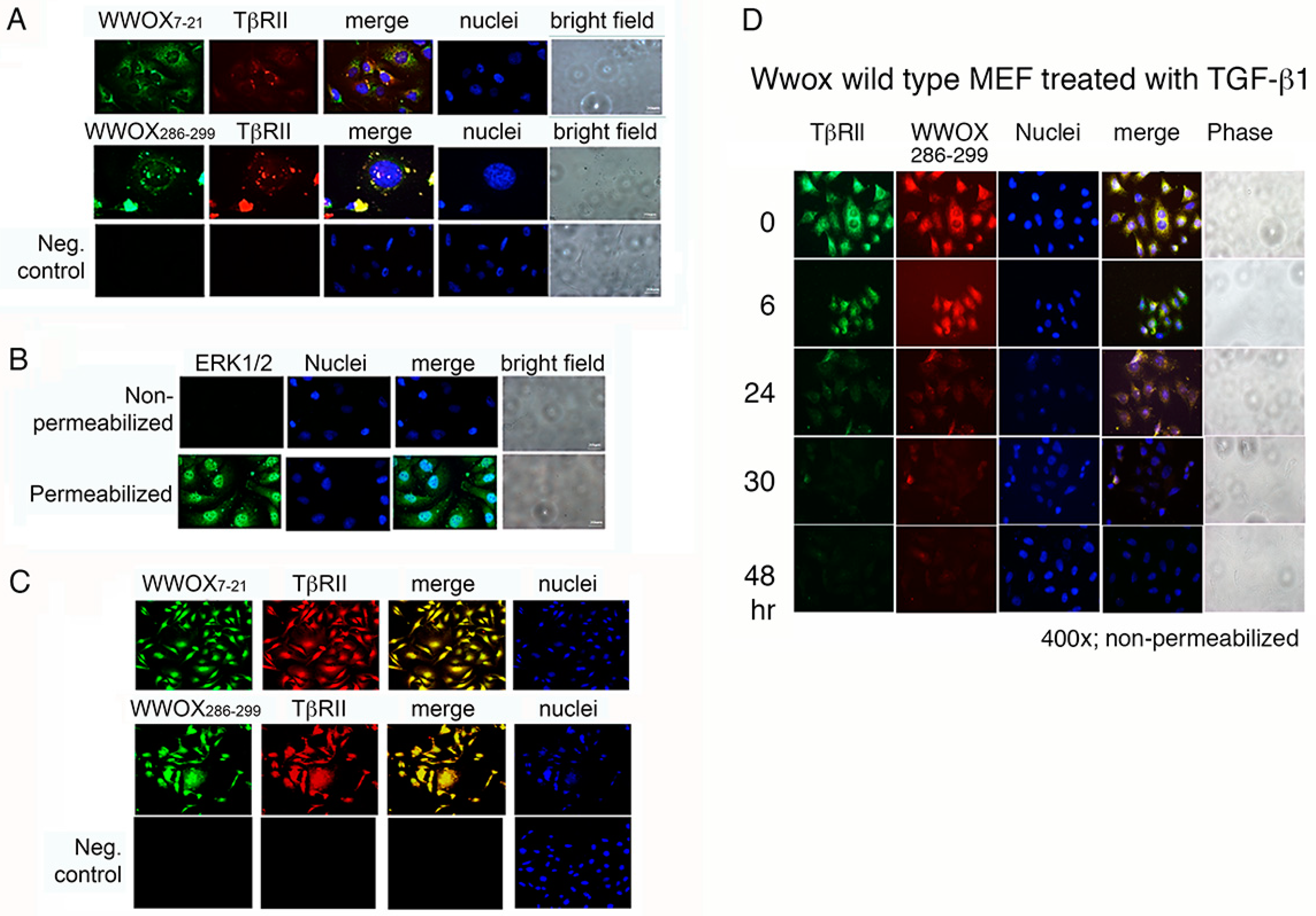
2.5. WWOX7-21 and WWOX286-299 Peptides Bind Cell Surface and Colocalize with Membrane Type II TGF-β Receptor (TβRII)
2.6. WWOX Is Clustered in the Cell Membrane
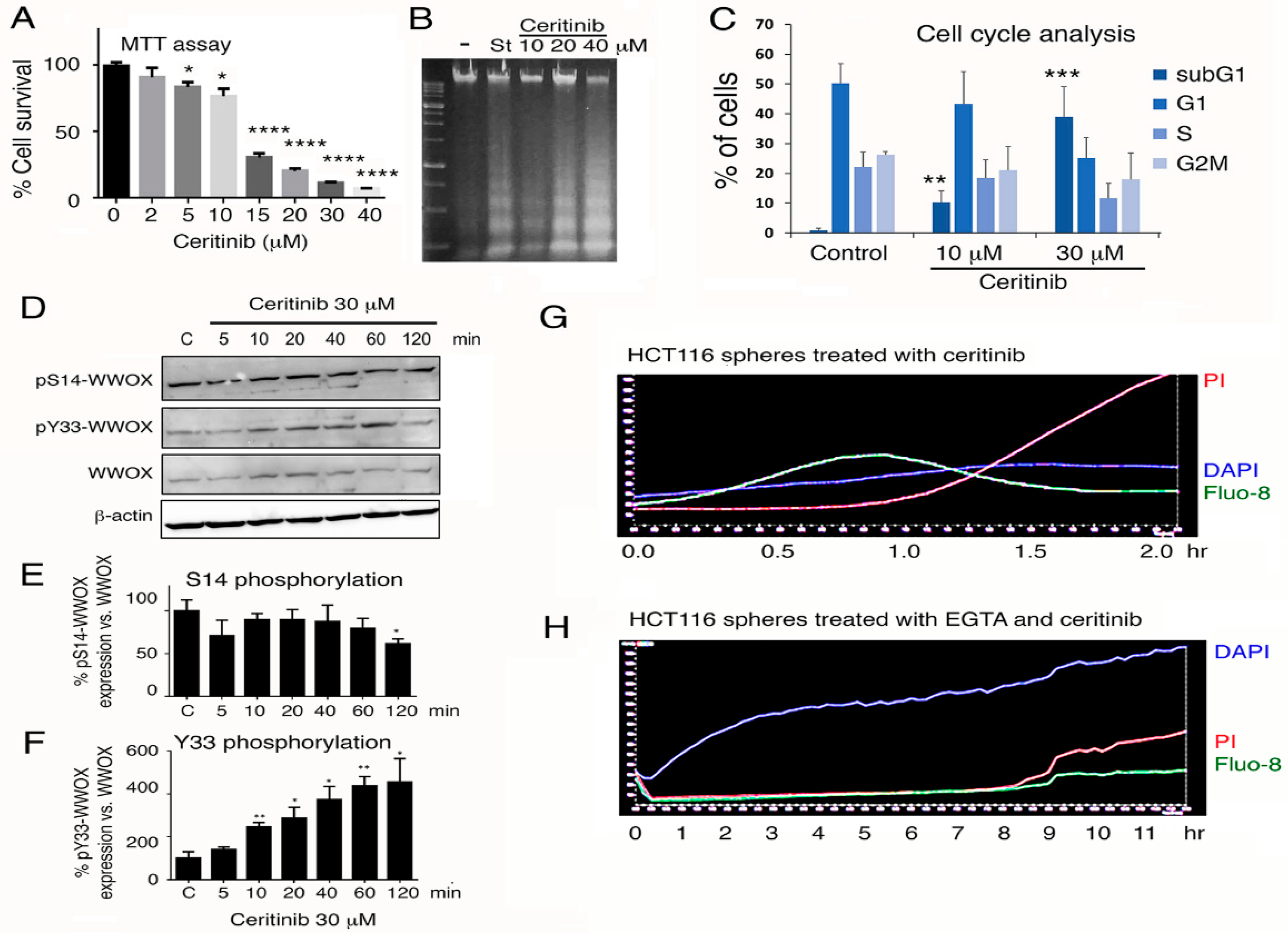
2.7. Ceritinib Mediates 4T1 Cell Sphere Shinkage (Pre-Explosion Stage) and then Explosion and Death (Explosion Stage)
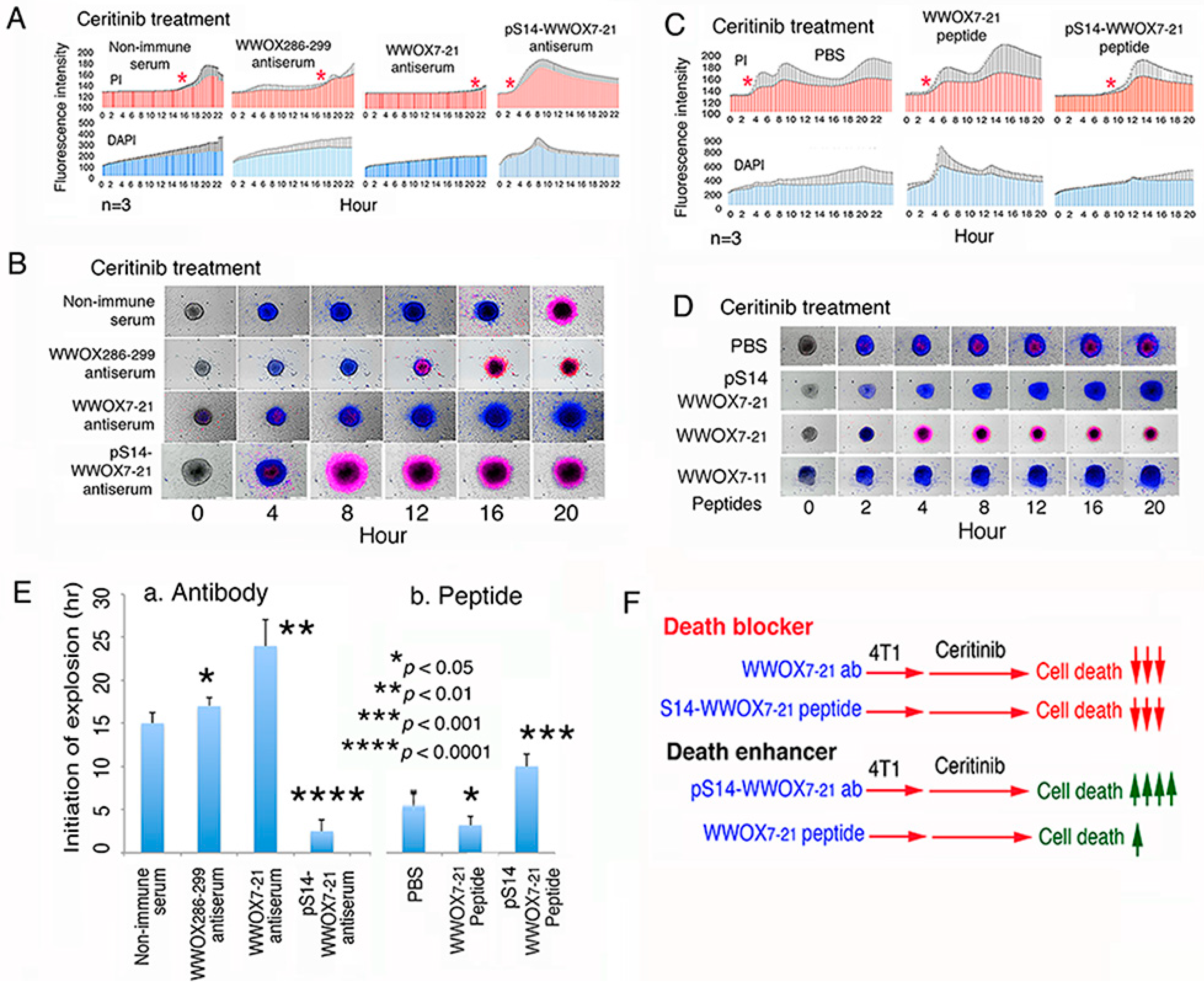
2.8. Treatment of 4T1 Cells with pS14-WWOX Antibody Accelerates Ceritinib-Mediated Sphere Explosion and Cell Death
2.9. pS14-WWOX Peptide Protects 4T1 Cells from Ceritinib-Mediated Death In Vitro
2.10. Ceritinib Upregulates Proapoptotic pY33-WWOX and Meanwhile Induces Ca2+ Influx for Leading to Apoptosis of 4T1 Cells
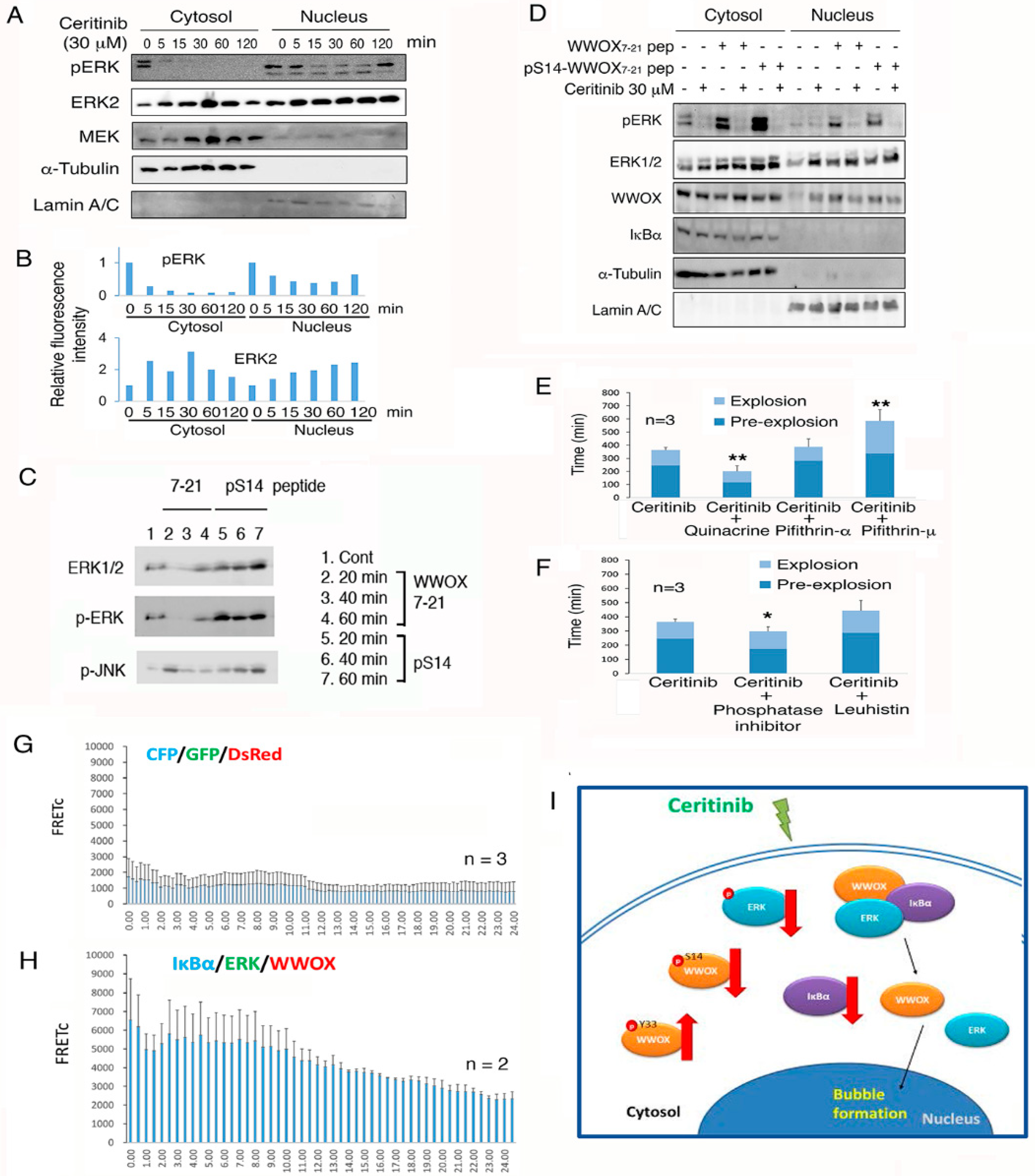
2.11. WWOX Peptides Counteract the Ceritinib-Mediated Apoptosis via Regulating ERK Phosphorylation
2.12. Endogenous p53 and Aminopeptidase M Enhance Ceritinib-Mediated Cell Sphere Explosion and Cell Death
2.13. Ceritinib Suppresses the Prosurvival IkBα/ERK/WWOX Signaling to Cause Cell Death
3. Discussion
3.1. pY33 Switching to pS14 for Cancer Promotion in WWOX
3.2. Role of TβRII in Anchoring WWOX Peptides
3.3. WWOX7-21 and pS14-WWOX7-21 Peptides Recapitulate the Functional Properties of Endogenous WWOX
3.4. Zfra Induces the Hyal-2/WWOX/Smad4 Signaling for Cancer Suppression
3.5. WWOX Peptides and Their Anticancer Activities in Immune Competent and Deficient Mice
3.6. pS14-WWOX7-21 Peptide Induces the Expansion of Spleen CD8α+ T and CD19+ B Cells
3.7. pS14-WWOX7-21 Peptide Probably Drives the IkBα/WWOX/ERK Signaling for T/B Cell Maturation
3.8. The Complex of pY33-WWOX and Hyal-2 Causes Apoptosis in the Nucleus
3.9. Ceritinib Upregulates pY33-WWOX, Downregulates p-ERK, Induces Ca+2 Influx, and Ultimately Generates DNA Fragmentation
4. Materials and Methods
4.1. Cell Lines
4.2. Structure Simulation and Peptide Synthesis
4.3. Cancer Growth and Immune Cell Differentiation in Mice
4.4. Antibodies, Immunohistochemistry, and Immunofluorescence Microscopy
4.5. Time-Lapse Microscopy for 4T1 Stem Cell Sphere Explosion and Death
4.6. Time-Lapse tri-Molecular Förster Resonance Energy Transfer (FRET) Microscopy
4.7. Data Presentation and Statistical Analysis
4.8. Ethics Approval in Animal Use
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bednarek, A.K.; Laflin, K.J.; Daniel, R.L.; Liao, Q.; Hawkins, K.A.; Aldaz, C.M. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3–24.1, a region frequently affected in breast cancer. Cancer Res. 2000, 60, 2140–2145. [Google Scholar] [PubMed]
- Ried, K.; Finnis, M.; Hobson, L.; Mangelsdorf, M.; Dayan, S.; Nancarrow, J.K.; Woollatt, E.; Kremmidiotis, G.; Gardner, A.; Venter, D. Common chromosomal fragile site FRA16D sequence: Identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 2000, 9, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; Pratt, N.; Heath, J.; Schultz, L.; Sleve, D.; Carey, G.B.; Zevotek, N. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J. Biol. Chem. 2001, 276, 3361–3370. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; Hsu, L.J.; Lin, Y.S.; Lai, F.J.; Sheu, H.M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef]
- Hsu, L.J.; Chiang, M.F.; Sze, C.I.; Su, W.P.; Yap, Y.V.; Lee, I.T.; Kuo, H.L.; Chang, N.S. HYAL-2-WWOX-SMAD4 Signaling in Cell Death and Anticancer Response. Front. Cell Dev. Biol. 2016, 4, 141. [Google Scholar] [CrossRef]
- Abu-Odeh, M.; Bar-Mag, T.; Huang, H.; Kim, T.; Salah, Z.; Abdeen, S.K.; Sudol, M.; Reichmann, D.; Sidhu, S.; Kim, P.M. Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J. Biol. Chem. 2014, 289, 8865–8880. [Google Scholar] [CrossRef]
- Aldaz, C.M.; Ferguson, B.W.; Abba, M.C. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim. Biophys. Acta (BBA) Rev. Cancer 2015, 1846, 188–200. [Google Scholar] [CrossRef]
- Chang, H.T.; Liu, C.C.; Chen, S.T.; Yap, Y.V.; Chang, N.S.; Sze, C.I. WW domain-containing oxidoreductase in neuronal injury and neurological diseases. Oncotarget 2014, 5, 11792–11799. [Google Scholar] [CrossRef]
- Alkhateeb, A.M.; Aburahma, S.K.; Habbab, W.; Thompson, I.R. Novel mutations in WWOX, RARS2, and C10orf2 genes in consanguineous Arab families with intellectual disability. Metab. Brain Dis. 2016, 31, 901–907. [Google Scholar] [CrossRef]
- Elsaadany, L.; El-Said, M.; Ali, R.; Kamel, H.; Ben-Omran, T. W44X mutation in the WWOX gene causes intractable seizures and developmental delay: A case report. BMC Med. Genet. 2016, 17, 53. [Google Scholar] [CrossRef]
- Tabarki, B.; Al Mutairi, F.; Al Hashem, A. The fragile site WWOX gene and the developing brain. Exp. Biol. Med. 2015, 240, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Sze, C.I.; Su, M.; Pugazhenthi, S.; Jambal, P.; Hsu, L.J.; Heath, J.; Schultz, L.; Chang, N.S. Down-regulation of WW domain-containing oxidoreductase induces tau phosphorylation in vitro a potential role in ALZHEIMER’S disease. J. Biol. Chem. 2004, 279, 30498–30506. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Ho, P.C.; Lee, I.T.; Chen, Y.A.; Chu, C.H.; Teng, C.C.; Wu, S.N.; Sze, C.I.; Chiang, M.F.; Chang, N.S. WWOX phosphorylation, signaling, and role in neurodegeneration. Front. Neurosci. 2018, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie Wolf, A. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.Y.; Lin, S.R.; Lee, M.H.; Schultz, L.; Sze, C.I.; Chang, N.S. A p53/TIAF1/WWOX triad exerts cancer suppression but may cause brain protein aggregation due to p53/WWOX functional antagonism. Cell Commun. Signal. 2019, 17, 76. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; Doherty, J.; Ensign, A.; Schultz, L.; Hsu, L.J.; Hong, Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J. Biol. Chem. 2005, 280, 43100–43108. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.S.; Doherty, J.; Ensign, A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J. Biol. Chem. 2003, 278, 9195–9202. [Google Scholar] [CrossRef]
- Lo, J.Y.; Chou, Y.T.; Lai, F.J.; Hsu, L.J. Regulation of cell signaling and apoptosis by tumor suppressor WWOX. Exp. Biol. Med. 2015, 240, 383–391. [Google Scholar] [CrossRef]
- Chen, S.J.; Lin, P.W.; Lin, H.P.; Huang, S.S.; Lai, F.J.; Sheu, H.M.; Hsu, L.J.; Chang, N.S. UV irradiation/cold shock-mediated apoptosis is switched to bubbling cell death at low temperatures. Oncotarget 2015, 6, 8007–8018. [Google Scholar] [CrossRef]
- Chang, N.S.; Schultz, L.; Hsu, L.J.; Lewis, J.; Su, M.; Sze, C.I. 17β-Estradiol upregulates and activates WOX1/WWOXv1 and WOX2/WWOXv2 in vitro: Potential role in cancerous progression of breast and prostate to a premetastatic state in vivo. Oncogene 2005, 24, 714–723. [Google Scholar] [CrossRef]
- Su, W.P.; Chen, S.H.; Chen, S.J.; Chou, P.Y.; Huang, C.C.; Chang, N.S. WW Domain-containing oxidoreductase is a potential receptor for sex steroid hormones. In Sex Hormones; Raghvendra, D., Ed.; InTech-Open Access Publisher: London, UK, 2012; pp. 333–351. [Google Scholar]
- Del Mare, S.; Husanie, H.; Iancu, O.; Abu-Odeh, M.; Evangelou, K.; Lovat, F.; Volinia, S.; Gordon, J.; Amir, G.; Stein, J. WWOX and p53 dysregulation synergize to drive the development of osteosarcoma. Cancer Res. 2016, 76, 6107–6117. [Google Scholar] [CrossRef] [PubMed]
- Abu-Odeh, M.; Salah, Z.; Herbel, C.; Hofmann, T.G.; Aqeilan, R.I. WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc. Natl. Acad. Sci. USA 2014, 111, E4716–E4725. [Google Scholar] [CrossRef] [PubMed]
- Abu-Remaileh, M.; Joy-Dodson, E.; Schueler-Furman, O.; Aqeilan, R.I. Pleiotropic functions of tumor suppressor WWOX in normal and cancer cells. J. Biol. Chem. 2015, 290, 30728–30735. [Google Scholar] [CrossRef] [PubMed]
- Schrock, M.S.; Batar, B.; Lee, J.; Druck, T.; Ferguson, B.; Cho, J.H.; Akakpo, K.; Hagrass, H.; Heerema, N.A.; Xia, F. Wwox–Brca1 interaction: Role in DNA repair pathway choice. Oncogene 2017, 36, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, L.V.; Lee, C.S.; Choo, A.; Richards, R.I. Tumor suppressor WWOX contributes to the elimination of tumorigenic cells in Drosophila melanogaster. PLoS ONE 2015, 10, e0136356. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Schultz, L.; Hong, Q.; Van Moer, K.; Heath, J.; Li, M.Y.; Lai, F.J.; Lin, S.R.; Lee, M.H.; Lo, C.P. Transforming growth factor β1 signaling via interaction with cell surface Hyal-2 and recruitment of WWOX/WOX1. J. Biol. Chem. 2009, 284, 16049–16059. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Hong, Q.; Chen, S.T.; Kuo, H.L.; Schultz, L.; Heath, J.; Lin, S.R.; Lee, M.H.; Li, D.Z.; Li, Z.L. Hyaluronan activates Hyal-2/WWOX/Smad4 signaling and causes bubbling cell death when the signaling complex is overexpressed. Oncotarget 2017, 8, 19137–19155. [Google Scholar] [CrossRef]
- Chou, P.Y.; Lai, F.J.; Chen, Y.A.; Sie, Y.D.; Kuo, H.L.; Su, W.P.; Wu, C.Y.; Liu, T.Y.; Wen, K.Y.; Hsu, L.J. Strategies by which WWOX-deficient metastatic cancer cells utilize to survive via dodging, compromising, and causing damage to WWOX-positive normal microenvironment. Cell Death Discov. 2019, 5, 97. [Google Scholar] [CrossRef]
- Lee, M.H.; Su, W.P.; Wang, W.J.; Lin, S.R.; Lu, C.Y.; Chen, Y.A.; Chang, J.Y.; Huang, S.S.; Chou, P.Y.; Ye, S.R. Zfra activates memory Hyal-2+ CD3−CD19−spleen cells to block cancer growth, stemness, and metastasis in vivo. Oncotarget 2015, 6, 3737–3751. [Google Scholar] [CrossRef]
- Lee, M.H.; Shih, Y.H.; Lin, S.R.; Chang, J.Y.; Lin, Y.H.; Sze, C.I.; Kuo, Y.M.; Chang, N.S. Zfra restores memory deficits in Alzheimer’s disease triple-transgenic mice by blocking aggregation of TRAPPC6AΔ, SH3GLB2, tau, and amyloid β, and inflammatory NF-κB activation. Alzheimers Dement. 2017, 3, 189–204. [Google Scholar] [CrossRef]
- Hong, Q.; Hsu, L.J.; Schultz, L.; Pratt, N.; Mattison, J.; Chang, N.S. Zfra affects TNF-mediated cell death by interacting with death domain protein TRADD and negatively regulates the activation of NF-κB, JNK1, p53 and WOX1 during stress response. BMC Mol. Biol. 2007, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.S.; Su, W.P.; Lin, H.P.; Kuo, H.L.; Wei, H.L.; Chang, N.S. Role of WW domain-containing oxidoreductase WWOX in driving T cell acute lymphoblastic leukemia maturation. J. Biol. Chem. 2016, 291, 17319–17331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.S.; Chang, N.S. Phosphorylation/de-phosphorylation in specific sites of tumor suppressor WWOX and control of distinct biological events. Exp. Biol. Med. 2018, 243, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Targeting the stem cell properties of adult breast cancer cells: Using combinatorial strategies to overcome drug resistance. Curr. Mol. Biol. Rep. 2017, 3, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Sneha, S.; Nagare, R.P.; Priya, S.K.; Sidhanth, C.; Pors, K.; Ganesan, T.S. Therapeutic antibodies against cancer stem cells: A promising approach. Cancer Immunol. Immunother. 2017, 66, 1383–1398. [Google Scholar] [CrossRef]
- Kuo, H.L.; Ho, P.C.; Huang, S.S.; Chang, N.S. Chasing the signaling run by tri-molecular time-lapse FRET microscopy. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef]
- Chang, N.S. Bubbling cell death: A hot air balloon released from the nucleus in the cold. Exp. Biol. Med. 2016, 241, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- De Pas, T.; Pala, L.; Catania, C.; Conforti, F. Molecular and clinical features of second-generation anaplastic lymphoma kinase inhibitors: Ceritinib. Future Oncol. 2017, 13, 2629–2644. [Google Scholar] [CrossRef]
- McAtee, C.O.; Barycki, J.J.; Simpson, M.A. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 2014, 123, 1–34. [Google Scholar]
- Chang, N.S.; Su, W.P. Modified Hyaluronan and Uses Thereof in Cancer Treatment. U.S. Patent 9,375,447, 28 June 2016. [Google Scholar]
- Chang, N.S.; Lu, C.Y.; Su, W.P.; Chen, Y.A.; Wang, W.J. Z Cells Activated by Zinc Finger-Like Protein and Uses Thereof in Cancer Treatment. U.S. Patent 20,150,329,824, 17 January 2017. [Google Scholar]
- Su, W.P.; Wang, W.J.; Sze, C.I.; Chang, N.S. Zfra induction of memory anticancer response via a novel immune cell. Oncoimmunology 2016, 5, e1213935. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.S. Transforming growth factor-β1 blocks the enhancement of tumor necrosis factor cytotoxicity by hyaluronidase Hyal-2 in L929 fibroblasts. BMC Cell Biol. 2002, 3, 8. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.-J.; Ho, P.-C.; Nagarajan, G.; Chen, Y.-A.; Kuo, H.-L.; Subhan, D.; Su, W.-P.; Chang, J.-Y.; Lu, C.-Y.; Chang, K.T.; et al. WWOX Possesses N-Terminal Cell Surface-Exposed Epitopes WWOX7-21 and WWOX7-11 for Signaling Cancer Growth Suppression and Prevention In Vivo. Cancers 2019, 11, 1818. https://doi.org/10.3390/cancers11111818
Wang W-J, Ho P-C, Nagarajan G, Chen Y-A, Kuo H-L, Subhan D, Su W-P, Chang J-Y, Lu C-Y, Chang KT, et al. WWOX Possesses N-Terminal Cell Surface-Exposed Epitopes WWOX7-21 and WWOX7-11 for Signaling Cancer Growth Suppression and Prevention In Vivo. Cancers. 2019; 11(11):1818. https://doi.org/10.3390/cancers11111818
Chicago/Turabian StyleWang, Wan-Jen, Pei-Chuan Ho, Ganesan Nagarajan, Yu-An Chen, Hsiang-Ling Kuo, Dudekula Subhan, Wan-Pei Su, Jean-Yun Chang, Chen-Yu Lu, Katarina T. Chang, and et al. 2019. "WWOX Possesses N-Terminal Cell Surface-Exposed Epitopes WWOX7-21 and WWOX7-11 for Signaling Cancer Growth Suppression and Prevention In Vivo" Cancers 11, no. 11: 1818. https://doi.org/10.3390/cancers11111818