Lipid Metabolic Reprogramming in Hepatocellular Carcinoma

Abstract
:1. Introduction
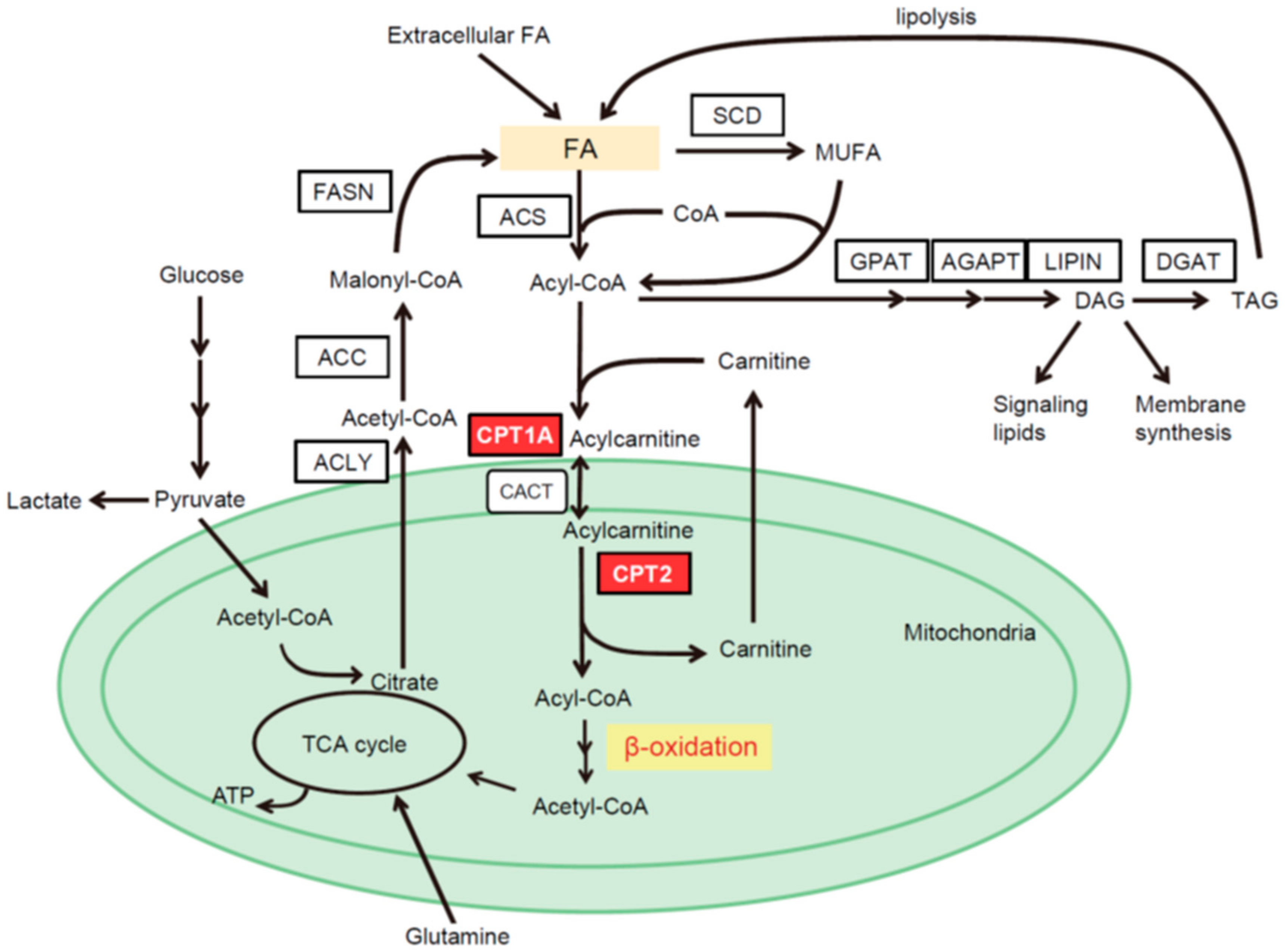
2. Intracellular Pathways of FA Metabolism
3. Roles of the FA Biosynthesis Pathway in HCC
3.1. ACC
3.2. FASN
3.3. SCD
3.4. SREBP-1
4. Lipid Metabolic Reprogramming in HCC in Response to Dyslipidemia Associated with Obesity or NASH
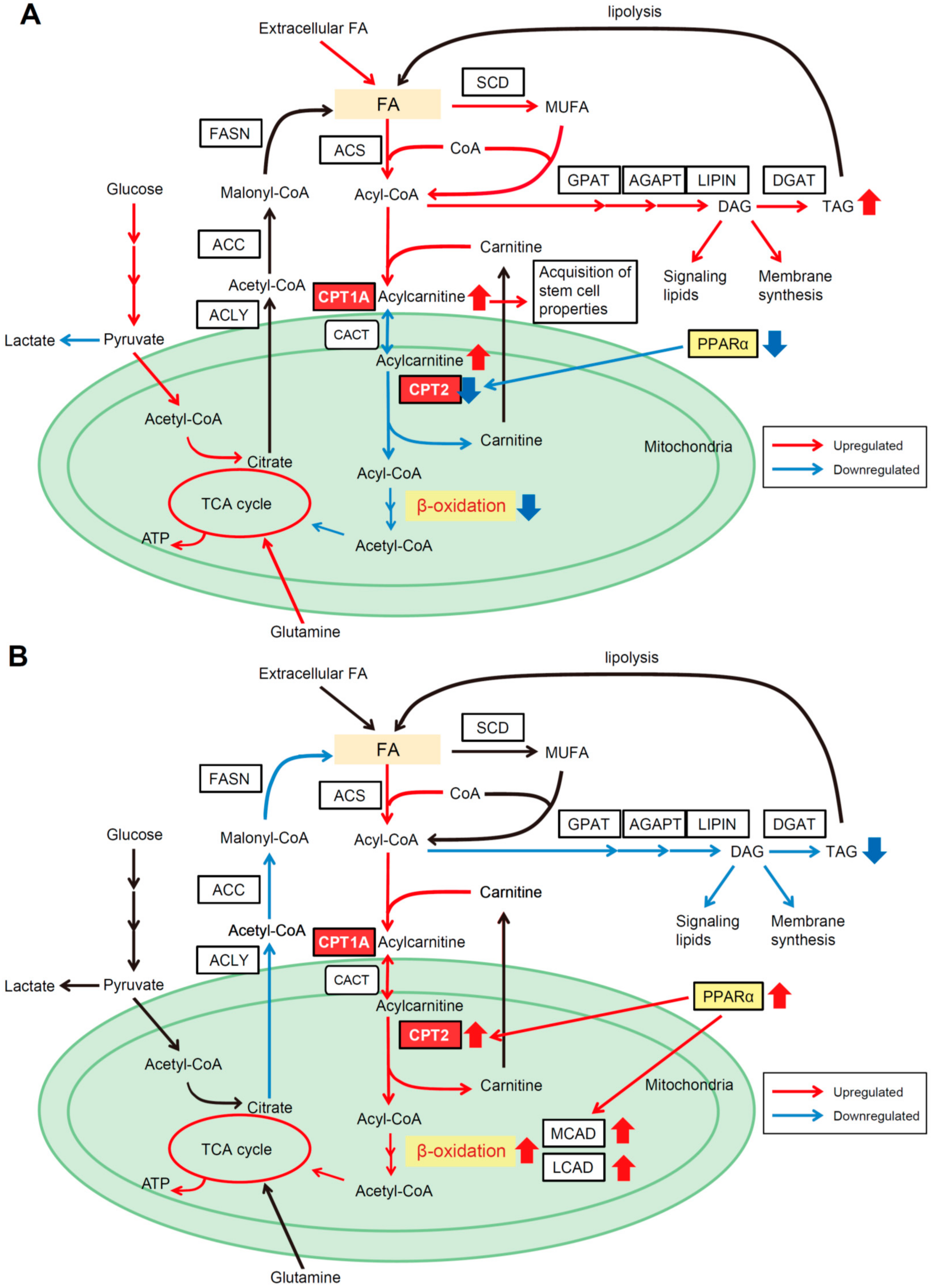
4.1. CPT2 Downregulation-Mediated Lipid Metabolic Reprogramming in Obesity- and NASH-Driven HCC
4.2. β-Catenin Determines the Dependence on FAO for HCC Development
5. Lipid Metabolic Reprogramming in HCC for Adaption to a Hypoxic Environment
6. Lipid Metabolic Reprogramming in Cancer Stem Cells
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Shiina, S.; Tateishi, R.; Arano, T.; Uchino, K.; Enooku, K.; Nakagawa, H.; Asaoka, Y.; Sato, T.; Masuzaki, R.; Kondo, Y.; et al. Radiofrequency ablation for hepatocellular carcinoma: 10-year outcome and prognostic factors. Am. J. Gastroenterol. 2012, 107, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Gluer, A.M.; Cocco, N.; Laurence, J.M.; Johnston, E.S.; Hollands, M.J.; Pleass, H.C.; Richardson, A.J.; Lam, V.W. Systematic review of actual 10-year survival following resection for hepatocellular carcinoma. HPB 2012, 14, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Kuk, D.; Gonen, M.; Balachandran, V.P.; Kingham, T.P.; Allen, P.J.; D’Angelica, M.I.; Jarnagin, W.R.; DeMatteo, R.P. Actual 10-year survivors after resection of hepatocellular carcinoma. Ann. Surg. Oncol. 2017, 24, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Maeda, S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J. Gastroenterol. 2012, 18, 4071–4081. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Tateishi, R.; Shiina, S.; Goto, E.; Sato, T.; Nakagawa, H.; Masuzaki, R.; Goto, T.; Hamamura, K.; Kanai, F.; et al. Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut 2009, 58, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Yang, H.I.; Yang, W.S.; Liu, C.J.; Chen, P.J.; You, S.L.; Wang, L.Y.; Sun, C.A.; Lu, S.N.; Chen, D.S.; et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: A follow-up study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Ohki, T.; Tateishi, R.; Sato, T.; Masuzaki, R.; Imamura, J.; Goto, T.; Yamashiki, N.; Yoshida, H.; Kanai, F.; Kato, N.; et al. Obesity is an independent risk factor for hepatocellular carcinoma development in chronic hepatitis C patients. Clin. Gastroenterol. Hepatol. 2008, 6, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H. How endoplasmic reticulum stress contributes to obesity-driven hepatic tumorigenesis. Hepat. Oncol. 2015, 2, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Arano, T.; Nakagawa, H.; Tateishi, R.; Ikeda, H.; Uchino, K.; Enooku, K.; Goto, E.; Masuzaki, R.; Asaoka, Y.; Kondo, Y.; et al. Serum level of adiponectin and the risk of liver cancer development in chronic hepatitis c patients. Int. J. Cancer 2011, 129, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IGA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H. Recent advances in mouse models of obesity- and nonalcoholic steatohepatitis-associated hepatocarcinogenesis. World J. Hepatol. 2015, 7, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Roessler, S.; Zhao, X.; Yu, Z.; Forgues, M.; Ji, J.; Karoly, E.; Qin, L.X.; Ye, Q.H.; Jia, H.L.; et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingold, J.A.; Zhu, D.; Lee, D.F.; Kaseb, A.; Chen, J. Genomic profiling and metabolic homeostasis in primary liver cancers. Trends Mol. Med. 2018, 24, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Nakagawa, H.; Enooku, K.; Kudo, Y.; Hayata, Y.; Nakatsuka, T.; Tanaka, Y.; Tateishi, R.; Hikiba, Y.; Misumi, K.; et al. Cpt2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 2018, 67, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P. Fatty acid metabolism and cancer development. Sci. Bull. 2016, 61, 1473–1479. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for srebp in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Bjornson, E.; Mukhopadhyay, B.; Asplund, A.; Pristovsek, N.; Cinar, R.; Romeo, S.; Uhlen, M.; Kunos, G.; Nielsen, J.; Mardinoglu, A. Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep. 2015, 13, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 2018, 155, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Wu, H.; Fu, G.B.; Zhang, H.L.; Zhou, X.; Tang, L.; Dong, L.W.; Qin, C.J.; Huang, S.; Zhao, L.H.; et al. Acetyl-coenzyme a carboxylase α promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology 2016, 63, 1272–1286. [Google Scholar] [CrossRef] [PubMed]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA carboxylase by phosphorylation or the inhibitor ND-654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.E.; Lahiri, S.; Chow, J.D.; Byrne, F.L.; Hargett, S.R.; Breen, D.S.; Olzomer, E.M.; Wu, L.E.; Cooney, G.J.; Turner, N.; et al. Inhibition of hepatic lipogenesis enhances liver tumorigenesis by increasing antioxidant defence and promoting cell survival. Nat. Commun. 2017, 8, 14689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Li, T.; Zhang, X.; Gao, P.; Qiao, P.; Li, S.; Geng, Z. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol. Rep. 2014, 32, 2471–2476. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.P.; Cheng, Z.L.; He, R.Y.; Song, L.; Tian, M.X.; Zhou, L.S.; Groh, B.S.; Liu, W.R.; Ji, M.B.; Ding, C.; et al. Destabilization of fatty acid synthase by acetylation inhibits de novo lipogenesis and tumor cell growth. Cancer Res. 2016, 76, 6924–6936. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular pathways: Fatty acid synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Tanaka, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Mohri, D.; Isomura, Y.; Seto, M.; Nakagawa, H.; Asaoka, Y.; et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J. Hepatol. 2011, 55, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M.; Sgroi, A.; Veyrat-Durebex, C.; Rubbia-Brandt, L.; Buhler, L.H.; Foti, M. Unsaturated fatty acids inhibit the expression of tumor suppressor phosphatase and tensin homolog (PTEN) via microRNA-21 up-regulation in hepatocytes. Hepatology 2009, 49, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.K.Y.; Kweon, S.M.; Chi, F.; Hwang, E.; Kabe, Y.; Higashiyama, R.; Qin, L.; Yan, R.; Wu, R.P.; Lai, K.; et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 2017, 152, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.K.F.; Lau, E.Y.T.; Leung, D.H.W.; Lo, J.; Ho, N.P.Y.; Cheng, L.K.W.; Ma, S.; Lin, C.H.; Copland, J.A.; Ding, J.; et al. Stearoyl-coa desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J. Hepatol. 2017, 67, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasegawa, K.; Ohashi, K.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur. J. Cancer 2005, 41, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Kamisuki, S.; Mao, Q.; Abu-Elheiga, L.; Gu, Z.; Kugimiya, A.; Kwon, Y.; Shinohara, T.; Kawazoe, Y.; Sato, S.; Asakura, K.; et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 2009, 16, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhou, Z.S.; Shen, Y.; Xu, J.; Miao, H.H.; Xiong, Y.; Xu, F.; Li, B.L.; Luo, J.; Song, B.L. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 2017, 65, 1936–1947. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qiu, L.; Wu, B.; Shen, H.; Zhu, J.; Zhou, L.; Gu, L.; Di, W. TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol. Med. Rep. 2013, 8, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Wheeler, F.B.; Fels, D.R.; Koumenis, C.; Kridel, S.J. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 2007, 67, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, Y.; Miyazaki, H.; Vaidyan, L.K.; Kagawa, Y.; Ebrahimi, M.; Yamamoto, Y.; Ogata, M.; Katsuyama, Y.; Sadahiro, H.; Suzuki, M.; et al. Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS ONE 2016, 11, e0147717. [Google Scholar] [CrossRef] [PubMed]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.; Caballeria, J.; Martinez-Arranz, I.; Dominguez-Diez, A.; Alonso, C.; Muntane, J.; Perez-Cormenzana, M.; Garcia-Monzon, C.; Mayo, R.; Martin-Duce, A.; et al. Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J. Proteome Res. 2012, 11, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Salomao, M.; Remotti, H.; Vaughan, R.; Siegel, A.B.; Lefkowitch, J.H.; Moreira, R.K. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum. Pathol. 2012, 43, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, J.; Ando, S.; Sakamoto, Y.; Kokudo, N.; Fukayama, M. Hepatocellular carcinoma with steatohepatitic features: A clinicopathological study of japanese patients. Histopathology 2014, 64, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Yaligar, J.; Teoh, W.W.; Othman, R.; Verma, S.K.; Phang, B.H.; Lee, S.S.; Wang, W.W.; Toh, H.C.; Gopalan, V.; Sabapathy, K.; et al. Longitudinal metabolic imaging of hepatocellular carcinoma in transgenic mouse models identifies acylcarnitine as a potential biomarker for early detection. Sci. Rep. 2016, 6, 20299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazanave, S.C.; Mott, J.L.; Elmi, N.A.; Bronk, S.F.; Werneburg, N.W.; Akazawa, Y.; Kahraman, A.; Garrison, S.P.; Zambetti, G.P.; Charlton, M.R.; et al. Jnk1-dependent puma expression contributes to hepatocyte lipoapoptosis. J. Biol. Chem. 2009, 284, 26591–26602. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A protein scaffold coordinates Src-mediated JNK activation in response to metabolic stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; Gao, P. Hif-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Lv, D.; Zheng, Y.; Wu, M.; Xu, C.; Zhang, Q.; Wu, L. Downregulation of cpt2 promotes tumorigenesis and chemoresistance to cisplatin in hepatocellular carcinoma. Onco. Targets Ther. 2018, 11, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Senni, N.; Savall, M.; Cabrerizo Granados, D.; Alves-Guerra, M.C.; Sartor, C.; Lagoutte, I.; Gougelet, A.; Terris, B.; Gilgenkrantz, H.; Perret, C.; et al. Beta-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Shibahara, J.; Hayashi, A.; Fukayama, M. β-catenin alteration is rare in hepatocellular carcinoma with steatohepatitic features: Immunohistochemical and mutational study. Virchows Arch. 2015, 467, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA. 2018, 115, E9879–E9888. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer lipid metabolism confers antiangiogenic drug resistance. Cell Metab. 2018, 28, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer stem cell metabolism and potential therapeutic targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, J.; Lima, J. Metabolic traits of cancer stem cells. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Int. 2017, 2017, 1656053. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Sacca, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Re, O.; Douet, J.; Buschbeck, M.; Fusilli, C.; Pazienza, V.; Panebianco, C.; Castracani, C.C.; Mazza, T.; Li Volti, G.; Vinciguerra, M. Histone variant macroh2a1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics 2018, 13, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Drugs | References |
|---|---|---|
| ACC (acetyl-CoA carboxylase) | ND-654, TOFA | [32,51] |
| FASN (fatty acid synthase) | TVB-2640, Cerulenin, Orlistat, C75, Triclosan, GSK2194069, Fasnall, EGCG | [40,41,52,53] |
| SCD (stearoyl-CoA desaturase) | A939572, CAY10566, MF-438, BZ36 | [54,55,56,57] |
| SREBP-1 (sterol regulatory element-binding protein) | Fatostatin, FGH10019 | [50,58] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2018, 10, 447. https://doi.org/10.3390/cancers10110447
Nakagawa H, Hayata Y, Kawamura S, Yamada T, Fujiwara N, Koike K. Lipid Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers. 2018; 10(11):447. https://doi.org/10.3390/cancers10110447
Chicago/Turabian StyleNakagawa, Hayato, Yuki Hayata, Satoshi Kawamura, Tomoharu Yamada, Naoto Fujiwara, and Kazuhiko Koike. 2018. "Lipid Metabolic Reprogramming in Hepatocellular Carcinoma" Cancers 10, no. 11: 447. https://doi.org/10.3390/cancers10110447