Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients

 , , and
, , and
Abstract
:1. Introduction
2. Results
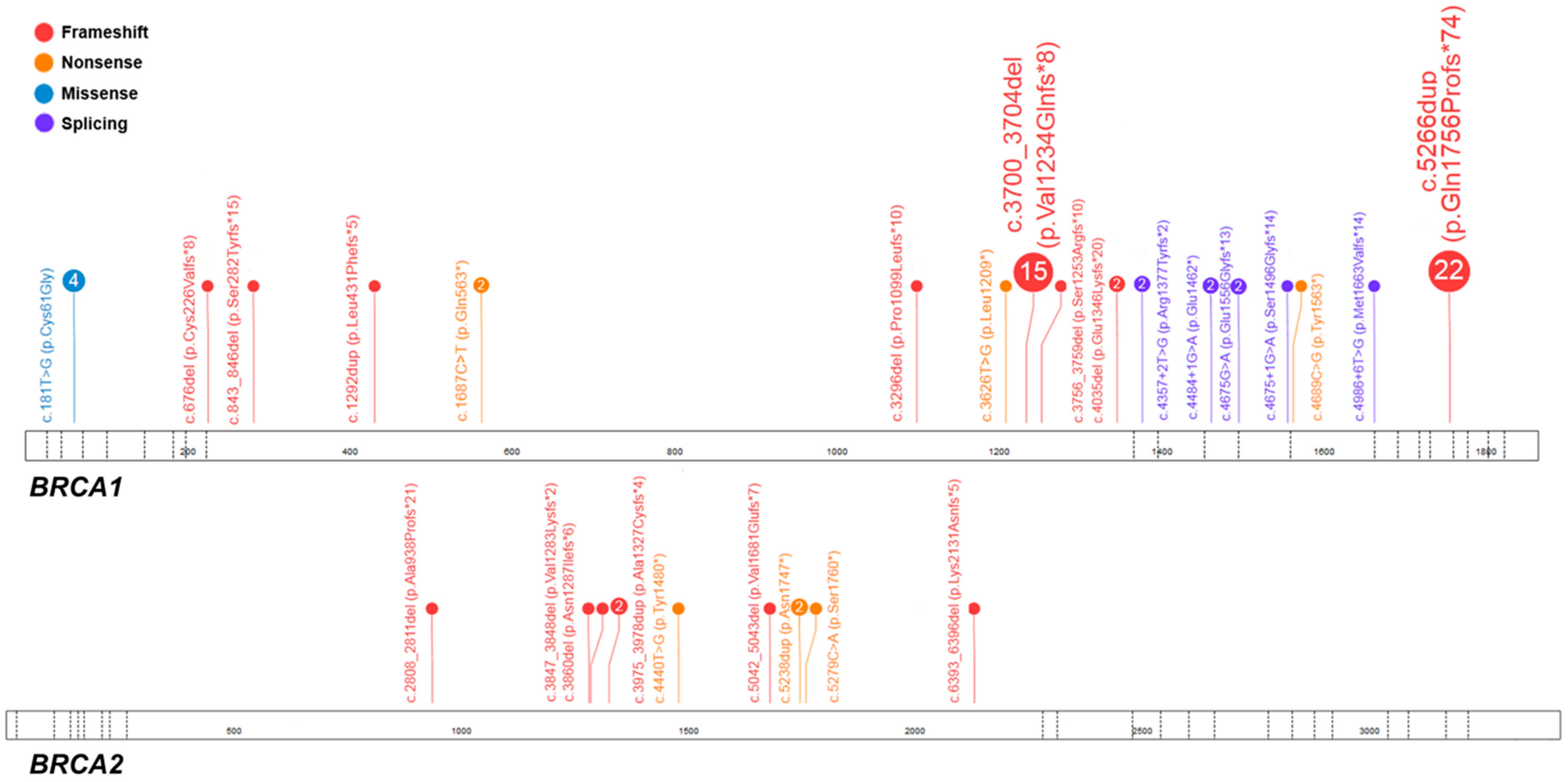
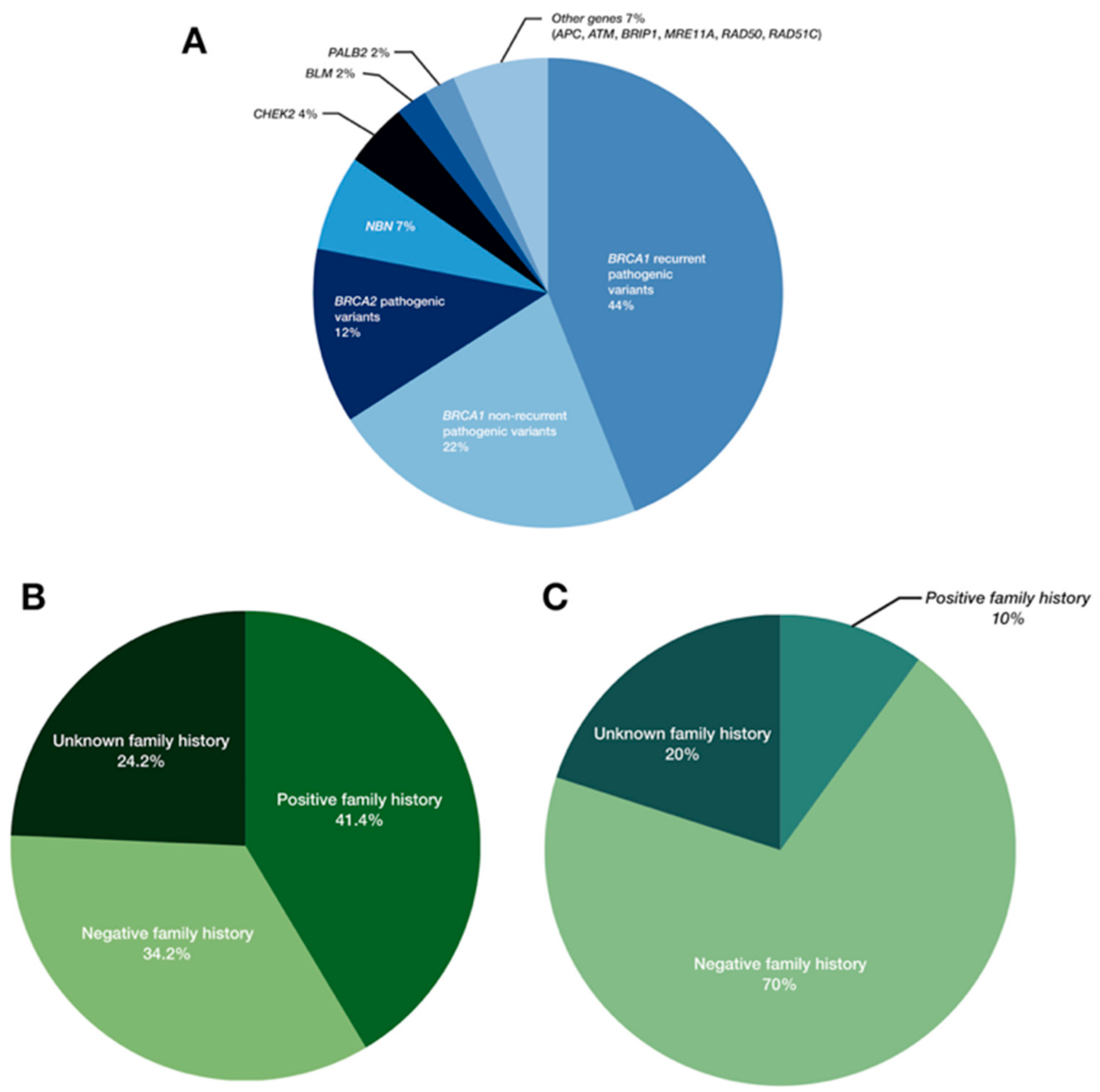
2.1. Pathogenic Variants in BRCA1/2
2.2. Pathogenic Variants in Moderate- and Low-Penetrance Genes
2.3. Variants of Uncertain Significance (VUS) in Low-Penetrance Genes
3. Discussion
4. Materials and Methods
4.1. Individuals and Sample Collection
4.2. DNA Extraction
4.3. Molecular Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, e359–e386. [Google Scholar] [CrossRef] [PubMed]
- Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; Chen, H.S.; et al. SEER Cancer Statistics Review, 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on April 2018).
- Farley, J.; Ozbun, L.L.; Birrer, M.J. Genomic analysis of epithelial ovarian cancer. Cell Res. 2008, 18, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012, 460, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Lee, M.K.; Casadei, S.; Thornton, A.M.; Stray, S.M.; Pennil, C.; Nord, A.S.; Mandell, J.B.; Swisher, E.M.; King, M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12629–12633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ratajska, M.; Krygier, M.; Stukan, M.; Kuzniacka, A.; Koczkowska, M.; Dudziak, M.; Sniadecki, M.; Debniak, J.; Wydra, D.; Brozek, I.; et al. Mutational analysis of BRCA1/2 in a group of 134 consecutive ovarian cancer patients. Novel and recurrent BRCA1/2 alterations detected by next generation sequencing. J. Appl. Genet. 2015, 56, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Brozek, I.; Cybulska, C.; Ratajska, M.; Piatkowska, M.; Kluska, A.; Balabas, A.; Dabrowska, M.; Nowakowska, D.; Niwinska, A.; Pamula-Pilat, J.; et al. Prevalence of the most frequent BRCA1 mutations in Polish population. J. Appl. Genet. 2011, 52, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozek, I.; Ochman, K.; Debniak, J.; Morzuch, L.; Ratajska, M.; Stepnowska, M.; Stukan, M.; Emerich, J.; Limon, J. High frequency of BRCA1/2 germline mutations in consecutive ovarian cancer patients in Poland. Gynecol. Oncol. 2008, 108, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Gorski, B.; Byrski, T.; Huzarski, T.; Jakubowska, A.; Menkiszak, J.; Gronwald, J.; Pluzanska, A.; Bebenek, M.; Fischer-Maliszewska, L.; Grzybowska, E.; et al. Founder mutations in the BRCA1 gene in Polish families with breast-ovarian cancer. Am. J. Hum. Genet. 2000, 66, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am. J. Hum. Genet. 2001, 68, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Kim, J.E.; Park, H.S.; Lee, S.T.; Park, J.S.; Han, J.W.; Lee, J.Y.; Kim, S.; Kim, S.W.; Kim, J.H.; et al. Detection of Germline Mutations in Patients with Epithelial Ovarian Cancer Using Multi-gene Panels: Beyond BRCA1/2. Cancer Res. Treat. 2018, 50, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minion, L.E.; Dolinsky, J.S.; Chase, D.M.; Dunlop, C.L.; Chao, E.C.; Monk, B.J. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol. 2015, 137, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women with Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet. Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Iniesta, M.D.; Gorin, M.A.; Chien, L.C.; Thomas, S.M.; Milliron, K.J.; Douglas, J.A.; Merajver, S.D. Absence of CHEK2*1100delC mutation in families with hereditary breast cancer in North America. Cancer Genet. Cytogenet. 2010, 202, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, K.M.; Bebel, A.; Vihinen, M.; Schleutker, J.; Sallinen, S.L. Screening for BRCA1, BRCA2, CHEK2, PALB2, BRIP1, RAD50, and CDH1 mutations in high-risk Finnish BRCA1/2-founder mutation-negative breast and/or ovarian cancer individuals. Breast Cancer Res. 2011, 13, e20. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. Biomed. Res. Int. 2015, 2015, e341723. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Nowakowska, D.; Niwinska, A.; Czapczak, D.; Kluska, A.; Piatkowska, M.; Wisniewska, A.; Paszko, Z. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int. J. Cancer 2006, 119, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet. Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef]
- Seemanova, E.; Sperling, K.; Neitzel, H.; Varon, R.; Hadac, J.; Butova, O.; Schrock, E.; Seeman, P.; Digweed, M. Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J. Med. Genet. 2006, 43, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Eccles, D.; Evans, D.G.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. J. Natl. Cancer Instig. 2015, 107, e214. [Google Scholar] [CrossRef] [PubMed]
- Anisimenko, M.S.; Kozyakov, A.E.; Paul, G.A.; Kovalenko, S.P. The frequency of the BLM p.Q548X (c.1642C>T) mutation in breast cancer patients from Russia is no higher than in the general population. Breast Cancer Res. Treat 2014, 148, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, N.; Togo, A.V.; Ratajska, M.; Kluzniak, W.; Takhirova, Z.; Tarp, T.; Prokofyeva, D.; Bermisheva, M.; Yanus, G.A.; Gorodnova, T.V.; et al. Prevalence of the BLM nonsense mutation, p.Q548X, in ovarian cancer patients from Central and Eastern Europe. Fam. Cancer 2015, 14, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Imyanitov, E.; Prokofyeva, D.; Bogdanova, N.; Dork, T. The frequency of the BLM*p.Q548X (c.1642C>T) mutation in breast cancer patients from Russia. Breast Cancer Res. Treat. 2014, 148, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Chui, M.H.; Ryan, P.; Radigan, J.; Ferguson, S.E.; Pollett, A.; Aronson, M.; Semotiuk, K.; Holter, S.; Sy, K.; Kwon, J.S.; et al. The histomorphology of Lynch syndrome-associated ovarian carcinomas: Toward a subtype-specific screening strategy. Am. J. Surg. Pathol. 2014, 38, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.; Permuth-Wey, J.; Kumar, A.; Sellers, T.A. Systematic review and meta-analysis of ovarian cancers: Estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin. Cancer Res. 2008, 14, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Andre, P.A.; Ratajska, M.; Kuzniacka, A.; Limon, J.; Tournier, B.B.; Colas, J.; Laurent, G.; Irminger-Finger, I. Antagonizing functions of BARD1 and its alternatively spliced variant BARD1delta in telomere stability. Oncotarget 2017, 8, 9339–9353. [Google Scholar] [CrossRef] [PubMed]
- Brozek, I.; Ratajska, M.; Piatkowska, M.; Kluska, A.; Balabas, A.; Dabrowska, M.; Nowakowska, D.; Niwinska, A.; Rachtan, J.; Steffen, J.; et al. Limited significance of family history for presence of BRCA1 gene mutation in Polish breast and ovarian cancer cases. Fam. Cancer 2012, 11, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dockery, L.E.; Gunderson, C.C.; Moore, K.N. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco. Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Case No. | Exon/Intron | Variant in Corresponding cDNA | Predicted Amino Acid Sequence | Variant Type | dbSNP ID 1 | ACMG Classification 2 | Age, Years | FIGO Stage | Histology | Family History |
|---|---|---|---|---|---|---|---|---|---|---|
| BRCA1 (NM_007294.3; LRG_292t1) | ||||||||||
| M115 | 5 | c.181T>G | p.(Cys61Gly) | M | rs28897672 | Pathogenic (PS3 + PS4 + PM2 + PP1+ PP5) | 36 | IIIC | serous | - |
| M296 | 54 | IIIC | serous | + | ||||||
| K199 | 46 | IIIC | serous | - | ||||||
| K221 | 51 | IIIC | serous | - | ||||||
| D166 | 11 | c.676del | p.(Cys226Valfs*8) | F | rs80357941 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 43 | ND | serous | - |
| K187 | 11 | c.843_846del | p.(Ser282Tyrfs*15) | F | rs80357919 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 46 | IIIC | serous | - |
| D18 | 11 | c.1292dup | p.(Leu431Phefs*5) | F | rs80357528 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 42 | IIIC | serous | + |
| D138 | 11 | c.1687C>T | p.(Gln563*) | N | rs80356898 | Pathogenic (PVS1+PS4+PM1+PM2 + PP1 + PP5) | 49 | ND | serous | ND |
| K100 | 45 | IIB | serous | + | ||||||
| M397 | 11 | c.3296del | p.(Pro1099Leufs*10) | F | rs80357815 | Pathogenic (PVS1 + PM2 + PM4 + PP5) | 59 | IIIC | serous | ND |
| D53 | 11 | c.3626T>G | p.(Leu1209*) | N | rs786203884 | Pathogenic (PVS1 + PS4 + PM2 + PM4 + PP1 + PP5) | 59 | ND | low differentiated | ND |
| M22 | 11 | c.3700_3704del | p.(Val1234Glnfs*8) | F | rs80357609 | Pathogenic (PVS1 + PM1 + PM2 + PP5) | 61 | IIIB | serous | ND |
| M38 | 43 | IIIC | serous | - | ||||||
| M66 | 52 | ND | serous | ND | ||||||
| D3 | 45 | IIIC | serous | + | ||||||
| D23 | 47 | IV | serous | - | ||||||
| D63 | 55 | IIIC | serous | + | ||||||
| D70 | 48 | ND | serous | ND | ||||||
| D71 | 47 | ND | serous | - | ||||||
| D104 | 56 | ND | low differentiated | - | ||||||
| D136 | 43 | IIIB | serous | - | ||||||
| D156 | 47 | ND | serous | - | ||||||
| K65 | 64 | IIIC | serous | + | ||||||
| K125 | 50 | IIIC | serous | - | ||||||
| K152 | 66 | IV | serous | + | ||||||
| K189 | 63 | IIIC | low differentiated | + | ||||||
| K74 | 11 | c.3756_3759del | p.(Ser1253Argfs*10) | F | rs80357868 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 45 | IV | serous | + |
| D15 | 11 | c.4035del | p.(Glu1346Lysfs*20) | F | rs80357711 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 54 | IIB | serous | - |
| K222 | ND | IIB | low differentiated | + | ||||||
| M92 | 13 | c.4357+2T>G; r.[4186_4357del];[=] | p.(Arg1377Tyrfs*2) | S | rs80358152 | Pathogenic (PVS1 + PS2 + PM2 + PP1 + PP3 + PP5) | 48 | IIIC | serous | + |
| M95 | 13 | 45 | IIIC | serous | - | |||||
| M395 | 14 | c.4484+1G>A; r.[4358_4484del];[=] | p.(Glu1462*) | S | rs80358063 | Pathogenic (PVS1 + PS3 + PP1 + PP3 + PP4 + PP5) | 41 | IIIC | serous | ND |
| K201 | 52 | IV | mesonephroid | + | ||||||
| D140 | 15 | c.4675G>A; r.[4665_4675del];[=] | p.(Glu1556Glyfs*13) | S | rs80356988 | Pathogenic (PS3 + PM1 + PM2 + PP1 + PP5) | 37 | IIIC | serous | ND |
| K73 K108 | 15 | c.4675+1G>A; r.[4485_4675del];[=] | p.(Ser1496Glyfs*14) | S | rs80358044 | Pathogenic (PVS1 + PS3 + PM1 + PM2 + PP1 + PP3 + PP5) | 44 41 | IIIC IIIC | serous clear cell | ND - |
| D152 | 16 | c.4689C>G | p.(Tyr1563*) | N | rs80357433 | Pathogenic (PVS1 + PS4 + PM1 + PP1 + PP3 + PP4) | 38 | ND | serous | - |
| D72 | 16 | c.4986+6T>G, r.[4916+1_4916+65ins];[=] | p.(Met1663Valfs*14) | S | rs80358086 | Pathogenic (PS3 + PS4 + PM1 + PM2 + PP1 + PP3 + PP5) | 47 | IIIC | serous | + |
| M50 | 20 | c.5266dup | p.(Gln1756Profs*74) | F | rs397507247 | Pathogenic (PVS1 + PM1 + PP5) | 52 | IIIC | serous | + |
| M108 | 63 | ND | serous | ND | ||||||
| M138 | 65 | ND | serous | + | ||||||
| M225 | 50 | IIID | serous | + | ||||||
| M226 | 51 | IIIC | serous | + | ||||||
| M227 | 58 | IIIC | serous | - | ||||||
| M314 | 46 | IIIC | endometrial | + | ||||||
| M323 | 37 | IIIC | serous | + | ||||||
| M368 | 60 | ND | serous | ND | ||||||
| M374 | 66 | ND | serous | ND | ||||||
| M378 | 36 | ND | serous | ND | ||||||
| D9 | 43 | IIIB | endometrial | + | ||||||
| D27 | 66 | IIIC | serous | ND | ||||||
| D66 | 47 | ND | serous | + | ||||||
| D83 | 50 | IIIC | serous | + | ||||||
| D99 | ND | IIIC | serous | ND | ||||||
| D105 | 50 | ND | serous | + | ||||||
| D144 | 56 | IIC | serous | ND | ||||||
| D149 | 40 | ND | serous | - | ||||||
| K53 | 60 | IIIC | serous | + | ||||||
| K121 | 52 | IIIB | clear cell | - | ||||||
| K197 | 43 | IIIC | endometrial | - | ||||||
| BRCA2(NM_000059.3; LRG_293t1) | ||||||||||
| M164 | 11 | c.2808_2811del | p.(Ala938Profs*21) | F | rs80359351 | Pathogenic (PVS1 + PS4 + PM2 + PP5) | 81 | ND | serous | + |
| D121 | 11 | c.3847_3848del | p.(Val1283Lysfs*2) | F | rs746229647 | Pathogenic (PVS1 + PM1 + PM2 + PP1 + PP5) | ND | ND | serous | ND |
| D114 | 11 | c.3860del | p.(Asn1287Ilefs*6) | F | rs80359406 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 54 | IIIC | serous | + |
| M8 | 11 | c.3975_3978dup | p.(Ala1327Cysfs*4) | F | rs764689249 | Pathogenic (PVS1 + PS4 + PM2 + PP5) | 45 | IV | serous | + |
| K183 | 56 | IV | low differentiated | + | ||||||
| K212 | 11 | c.4440T>G | p.(Tyr1480*) | N | rs397507719 | Pathogenic (PVS1 + PM1 + PM2 + PP1 + PP3 + PP5) | 54 | IIIC | serous | + |
| M178 | 11 | c.5042_5043del | p.(Val1681Glufs*7) | F | rs80359478 | Pathogenic (PVS1 + PS4 + PM1 + PM2 + PP1 + PP5) | 62 | IIIC | serous | - |
| D160 | 11 | c.5238dup | p.(Asn1747*) | F | rs80359499 | Pathogenic (PVS1 + PM1 + PM2 + PP1 + PP5) | 51 | IIIC | serous | - |
| K172 | 55 | IIIC | serous | - | ||||||
| K254 | 11 | c.5279C>A | p.(Ser1760*) | N | novel | Pathogenic (PVS1 + PM1 + PM2 + PP1 + PP3 + PP5) | ND | IV | mesonephroid | - |
| K93 | 11 | c.6393_6396del | p.(Lys2131Asnfs*5) | F | rs397507849 | Pathogenic (PVS1 + PM1 + PM2 + PP1 + PP5) | 76 | IIIC | serous | ND |
| Case No. | Exon/Intron | Variant in Corresponding cDNA | Predicted Amino Acid Sequence | Variant Type | dbSNP ID 1 | ACMG Classification 2 | Age, Years | FIGO Stage | Histology | Family History | BRCA1/2 Status |
|---|---|---|---|---|---|---|---|---|---|---|---|
| APC (NM_000038.4; LRG_130t1) | |||||||||||
| M33 | 16 | c.7927_7928del | p.(Leu2643Asnfs*10) | F | novel | Pathogenic (PVS1 + PM1 + PM2) | 41 | IIIC | serous | - | - |
| ATM(NM_000051.3, LRG_135t1) | |||||||||||
| D73 | 41 | c.6095G>A | p.(Arg2032Lys) | M | rs139770721 | Pathogenic (PS3 + PM2 + PP3 + PP5) | 59 | IC | serous/mucinous | - | - |
| BLM(NM_000057.2, LRG_20t1) | |||||||||||
| M66 | 2 | c.98 + 1G>C | p.(?) | S | rs750293380 | Pathogenic (PVS1 + PM2 + PP3) | 52 | ND | serous | - | BRCA1: c.3700_3704del |
| M71 | 7 | c.1642C>T | p.(Gln548*) | N | rs200389141 | Pathogenic (PVS1 + PP3 + PP5) | 61 | IIIC | serous | - | - |
| BRIP1 (NM_032043.2; LRG_300t1) | |||||||||||
| K190 | 11 | c.1510dup | p.(Ile504Asnfs*7) | F | rs775735278 | Pathogenic (PVS1 + PM2 + PP5) | 54 | IIB | endometrial | - | - |
| CHEK2(NM_007194.3) | |||||||||||
| M140 | 3 | c.444 + 1G>A, r.[ = ,444 + 1_444 + 2insATAG];[=] | p.(Glu149Ilefs*41) | S | rs121908698 | Pathogenic (PVS1 + PS3 + PP3 + PP5) | 54 | ND | myxoid leiomyosarcoma | ND | - |
| M374 | 11 | c.1100del | p.(Thr367Metfs*15) | F | rs555607708 | Pathogenic (PVS1 + PS3 + PP3 + PP5) | 66 | IIIC | serous | ND | BRCA1: c.5266dup |
| D104 | 56 | ND | low differentiated | - | BRCA1: c.3700_3704del | ||||||
| M113 | 11 | c.1169A>C | p.(Tyr390Ser) | M | rs200928781 | Likely pathogenic (PS3 + PM1 + PP3 + PP5) | 27 | IIIB | serous | - | - |
| MRE11A(NM_005591.3; LRG_85) | |||||||||||
| M42 | 3 | c.77T>C | p.(Met26Thr) | M | rs372068015 | Likely pathogenic (PM2 + PM3 + PP1 + PP5) | 61 | IIB | serous | - | - |
| NBN (NM_002485.4; LRG_158t1) | |||||||||||
| K74 | 4 | c.373A>T | p.(Lys125*) | N | novel | Pathogenic (PVS1 + PM2 + PP3) | 45 | IV | serous | + | BRCA1: c.3756_3759del |
| K124 | 6 | c.643C>T | p.(Arg215Trp) | M | rs34767364 | Likely pathogenic (PS2 + PM1 + PM3) | 47 | IIIC | serous | - | - |
| M86 | 6 | c.657_661del | p.(Lys219Asnfs*16) | F | rs587776650 | Pathogenic (PVS1 + PS3 + PM2 + PP5) | 64 | IIIC | serous | - | - |
| D131 | 76 | ND | serous | ND | - | ||||||
| K135 | 55 | IIIC | serous | - | - | ||||||
| K208 | ND | IIIB | serous | - | - | ||||||
| PALB2 (NM_024675.3; LRG_308t1) | |||||||||||
| M407 | 5 | c.2456_2463del | p.(Lys819Thrfs*4) | F | novel | Pathogenic (PVS1 + PM2 + PP5) | 53 | IIB | serous | ND | - |
| K219 | 7 | c.2632G>T | p.(Glu878*) | N | novel | Pathogenic (PVS1 + PM1 + PM2 + PP3 + PP5) | 54 | IIIC | ND | - | - |
| RAD50 (NM_005732.3) | |||||||||||
| M161 | 21 | c.3266_3273delinsT | p.(Lys1089Ilefs*17) | F | novel | Pathogenic (PVS1 + PM2 + PP5) | 46 | IA | serous | - | - |
| RAD51C(NM_058216.1; LRG_314t1) | |||||||||||
| K237 | 4 | c.706-2A>G, r.[706_837del];[=] | p.(Arg237_Val280del) | S | rs587780259 | Pathogenic (PVS1 + PM2 + PP3 + PP5) | ND | IIC | serous | + | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. https://doi.org/10.3390/cancers10110442
Koczkowska M, Krawczynska N, Stukan M, Kuzniacka A, Brozek I, Sniadecki M, Debniak J, Wydra D, Biernat W, Kozlowski P, et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers. 2018; 10(11):442. https://doi.org/10.3390/cancers10110442
Chicago/Turabian StyleKoczkowska, Magdalena, Natalia Krawczynska, Maciej Stukan, Alina Kuzniacka, Izabela Brozek, Marcin Sniadecki, Jaroslaw Debniak, Dariusz Wydra, Wojciech Biernat, Piotr Kozlowski, and et al. 2018. "Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients" Cancers 10, no. 11: 442. https://doi.org/10.3390/cancers10110442