
A Stand-Alone Microfluidic Chip for Long-Term Cell Culture
 , and
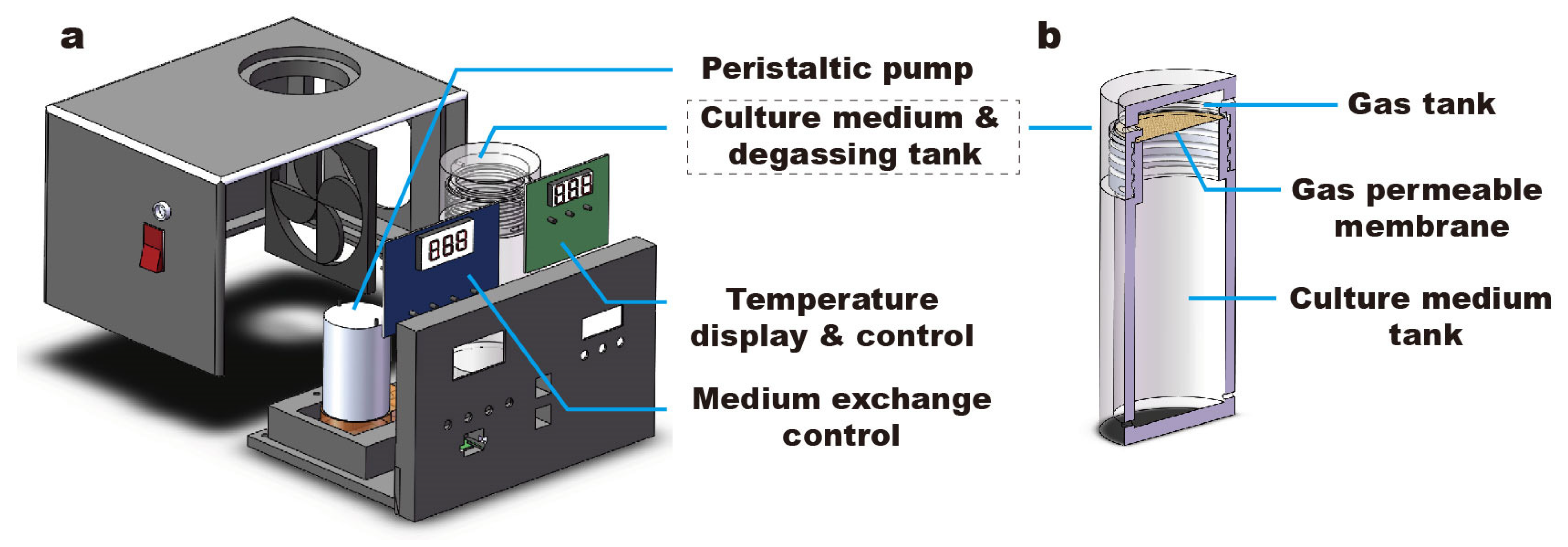
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Fabrication of Chip
2.2. Chip Control and Operation
2.3. Cell Culture
2.4. Cell Proliferation and Statistical Analysis
2.5. Numerical Simulation
3. Results
3.1. Parameter Determination of the Fluidic-Culture Device
3.2. Performance of the Fluidic-Cell-Culture Device
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Drew, J.; Machesky, L. The liver metastatic niche: Modelling the extracellular matrix in metastasis. Dis. Model. Mech. 2021, 14, dmm048801. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Gest, H. Homage to Robert Hooke (1635–1703): New insights from the recently discovered Hooke Folio. Perspect. Biol. Med. 2009, 52, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Hyllner, J.; Mason, C.; Wilmut, I. Cells: From Robert Hooke to cell therapy—A 350 year journey. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150320. [Google Scholar] [CrossRef] [Green Version]
- Che, B.; Zhao, W.; Liu, Y. Dynamic intracellular mechanical cues facilitate collective signaling responses. iScience 2021, 24, 102396. [Google Scholar] [CrossRef]
- Ettinger, A.; Wittmann, T. Fluorescence live cell imaging. Methods Cell Biol. 2014, 123, 77–94. [Google Scholar]
- Jensen, E.C. Overview of live-cell imaging: Requirements and methods used. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2013, 296, 1–8. [Google Scholar] [CrossRef]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [Green Version]
- Chudakov, D.; Matz, M.; Lukyanov, K. Fluorescent proteins and their applications in imaging living cells and tissues. Phyiological Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Jalili-Firoozinezhad, S.; Miranda, C. Modeling the human body on microfluidic chips. Trends Biotechnol. 2021, 39, 838–852. [Google Scholar] [CrossRef]
- Peel, S.; Corrigan, A.; Ewart, L. Introducing an automated high content confocal imaging approach for Organs-on-Chips. Lab A Chip 2019, 19, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.; Chung, B. An integrated microfluidic culture device to regulate endothelial cell differentiation from embryonic stem cells. Electrophoresis 2011, 32, 3133–3137. [Google Scholar] [CrossRef] [PubMed]
- Nocera, G.; Viscido, G.; Bernardo, D. The VersaLive platform enables microfluidic mammalian cell culture for versatile applications. Commun. Biol. 2022, 5, 1034. [Google Scholar] [CrossRef] [PubMed]
- Babic, J.; Griscom, L.; Coudreuse, D. An easy-to-build and re-usable microfluidic system for live-cell imaging. BMC Cell Biol. 2018, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, A.; Lopez, S.; Garnelo, S. Effect of extender, storage time and temperature on kinetic parameters (CASA) on bull semen samples. Biology 2021, 10, 806. [Google Scholar] [CrossRef]
- Gao, D.; Ma, Z.; Jiang, Y. Recent advances in microfluidic devices for foodborne pathogens detection. Trends Anal. Chem. 2022, 157, 116788. [Google Scholar] [CrossRef]
- Movizzo, P.; Ruder, W.; Long, Z. A 3D-printed portable device for field deployment of living biosensors. MethodsX 2019, 6, 1331–1335. [Google Scholar] [CrossRef]
- Sangal, A.; Pasini, P.; Daunert, S. Stability of spore-based biosensing systems under extreme conditions. Sens. Actuators B Chem. 2011, 158, 377–382. [Google Scholar] [CrossRef]
- Wang, J.; Maw, M.; Jiang, Z. Applications and perspectives on microfluidic technologies in ships and marine engineering: A review. Microfludics Nanofluidics 2017, 21, 39. [Google Scholar] [CrossRef]
- Arumugam, A.; Markham, C.; Aykar, S.S. PrintrLab incubator: A portable and low-cost CO2 incubator based on an open-source 3D printer architecture. PLoS ONE 2021, 16, e0251812. [Google Scholar] [CrossRef] [PubMed]
- Ham, R.G.; Puck, T. A regulated incubator controlling CO2 concentration, humidity and temperature for use in animal cell culture. Proc. Soc. Exp. Biol. Med. 1962, 111, 67–71. [Google Scholar] [CrossRef]
- Ince, C.; Ypey, D.L.; Diesselhoff-Den Dulk, M.M.; Visser, J.A.; De Vos, A.; Van Furth, R. Micro-CO2-incubator for use on a microscope. J. Immunol. Methods 1983, 60, 269–275. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, B.; Tian, Y.; Chen, H.; Liu, Y.; Fan, H.; Wang, K.; Zhang, C. Active fluidic chip produced using 3D-printing for combinatorial therapeutic screening on liver tumor spheroid. Biosens. Bioelectron. 2020, 151, 111966. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Y.; Zeng, Y.; Fu, J.; Che, B.; Jing, G.; Liu, Y.; Sun, D.; Zhang, C. A Stand-Alone Microfluidic Chip for Long-Term Cell Culture. Micromachines 2023, 14, 207. https://doi.org/10.3390/mi14010207
Feng Y, Zeng Y, Fu J, Che B, Jing G, Liu Y, Sun D, Zhang C. A Stand-Alone Microfluidic Chip for Long-Term Cell Culture. Micromachines. 2023; 14(1):207. https://doi.org/10.3390/mi14010207
Chicago/Turabian StyleFeng, Yibo, Yang Zeng, Jiahao Fu, Bingchen Che, Guangyin Jing, Yonggang Liu, Dan Sun, and Ce Zhang. 2023. "A Stand-Alone Microfluidic Chip for Long-Term Cell Culture" Micromachines 14, no. 1: 207. https://doi.org/10.3390/mi14010207