
Modelling Human Physiology on-Chip: Historical Perspectives and Future Directions

Abstract
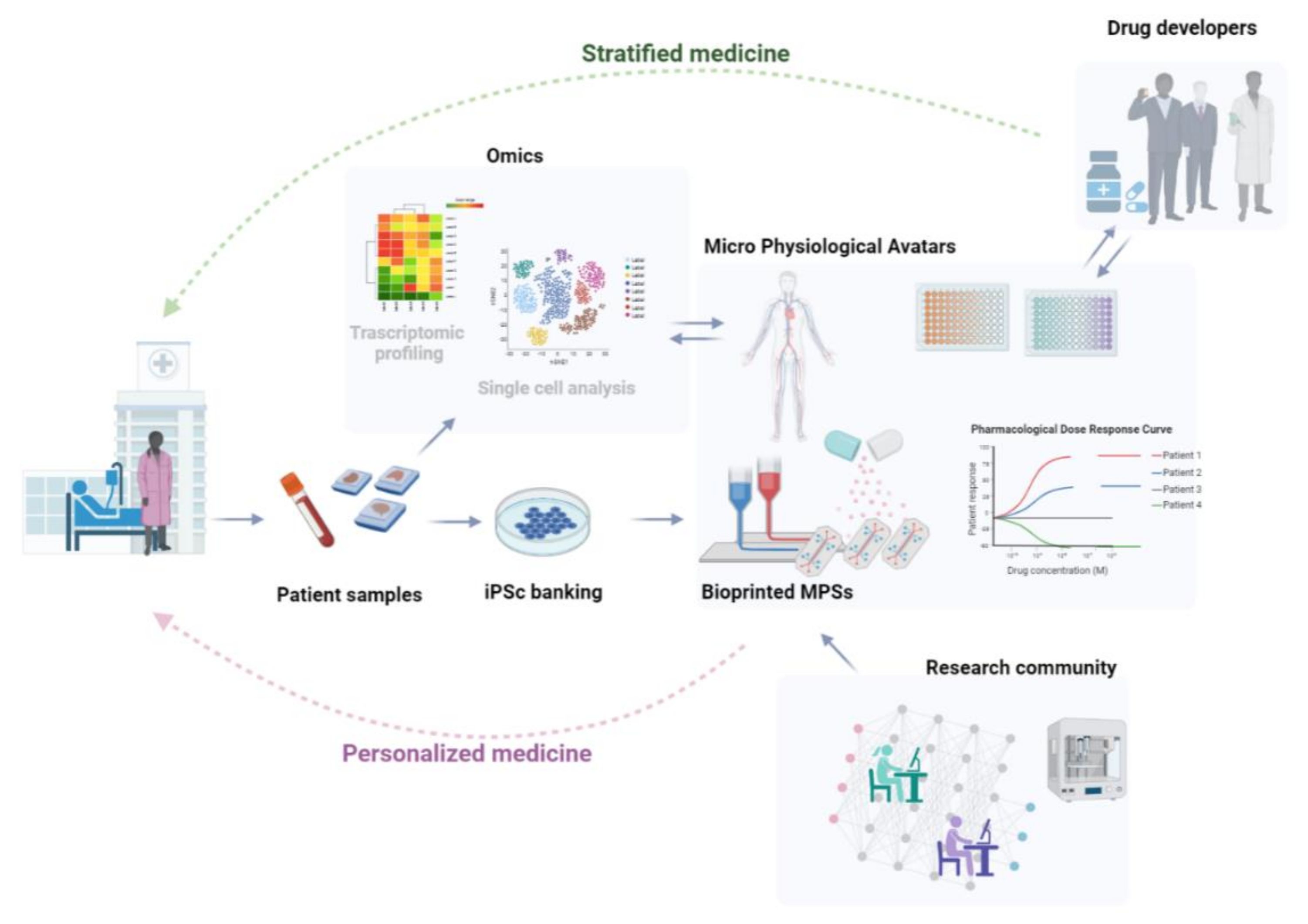
:1. Introduction
Terminology
2. Modelling Human Tissue Microenvironment in 2D and 3D Cell Cultures
2.1. Conventional 2D Models
2.2. 3D Models: Spheroids and Organoids
3. Dynamic Control of the Cell Microenvironment
3.1. Mechanobiology: From a Descriptive Field to Experimental Science
3.2. Cells and Tissues on-Chip
3.3. The First Organ-on-a-Chip
4. Synergistic Partnerships between Industry and Regulatory Bodies Are Driving Broad Adoption of OOCs and MPSs
5. Prominent Chip-Designs and Evolution of Technological Trajectories
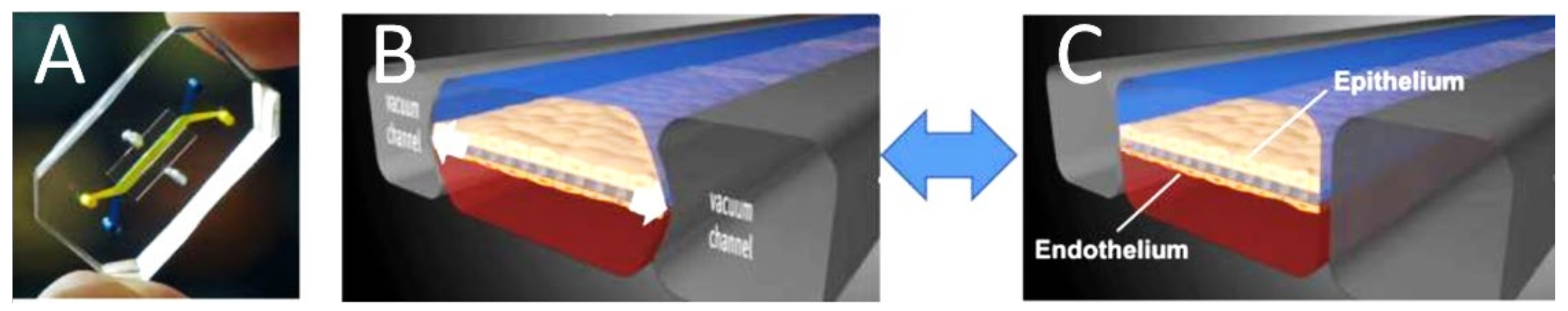
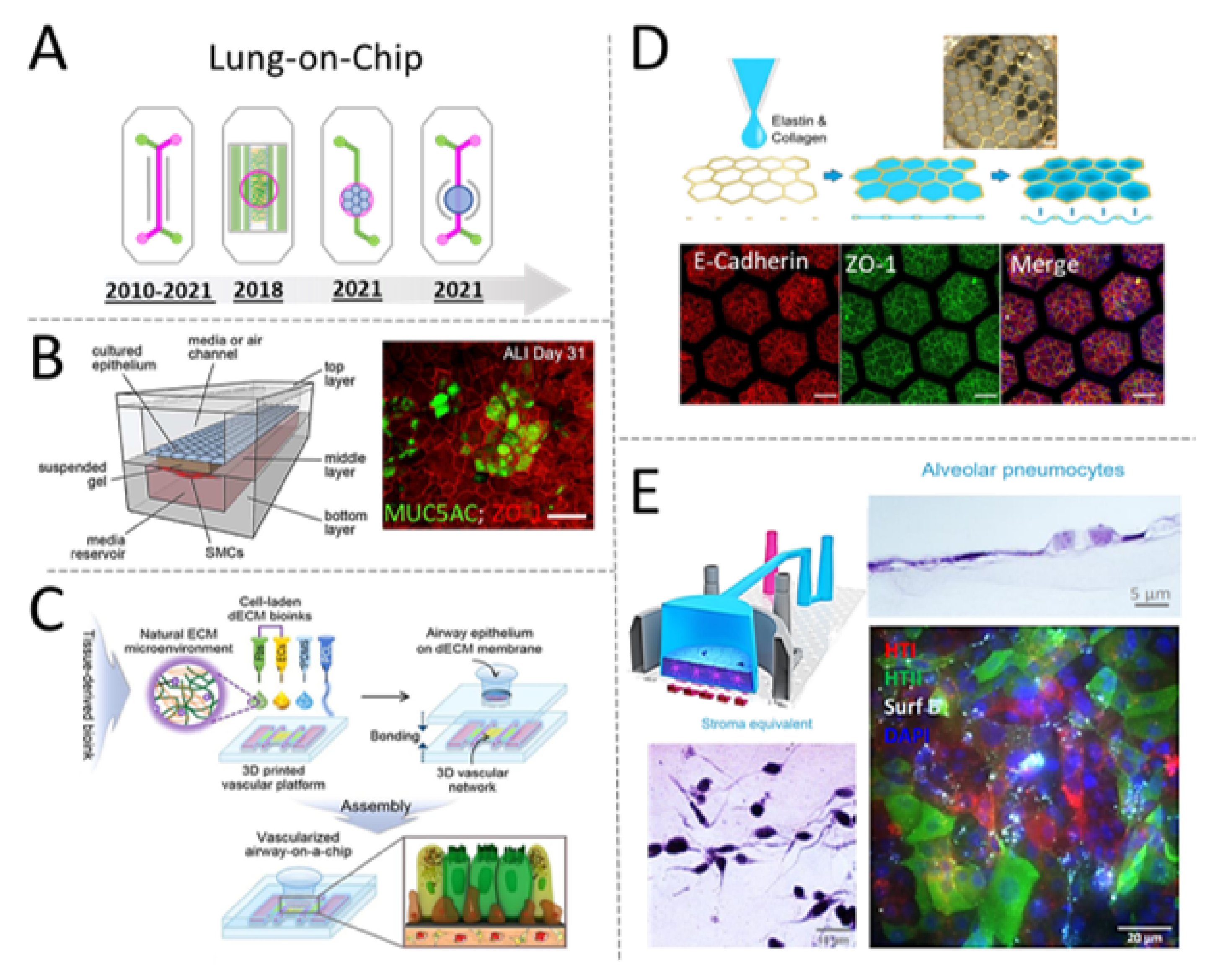
5.1. Tracing the Design Evolution of the Lung-on-Chip

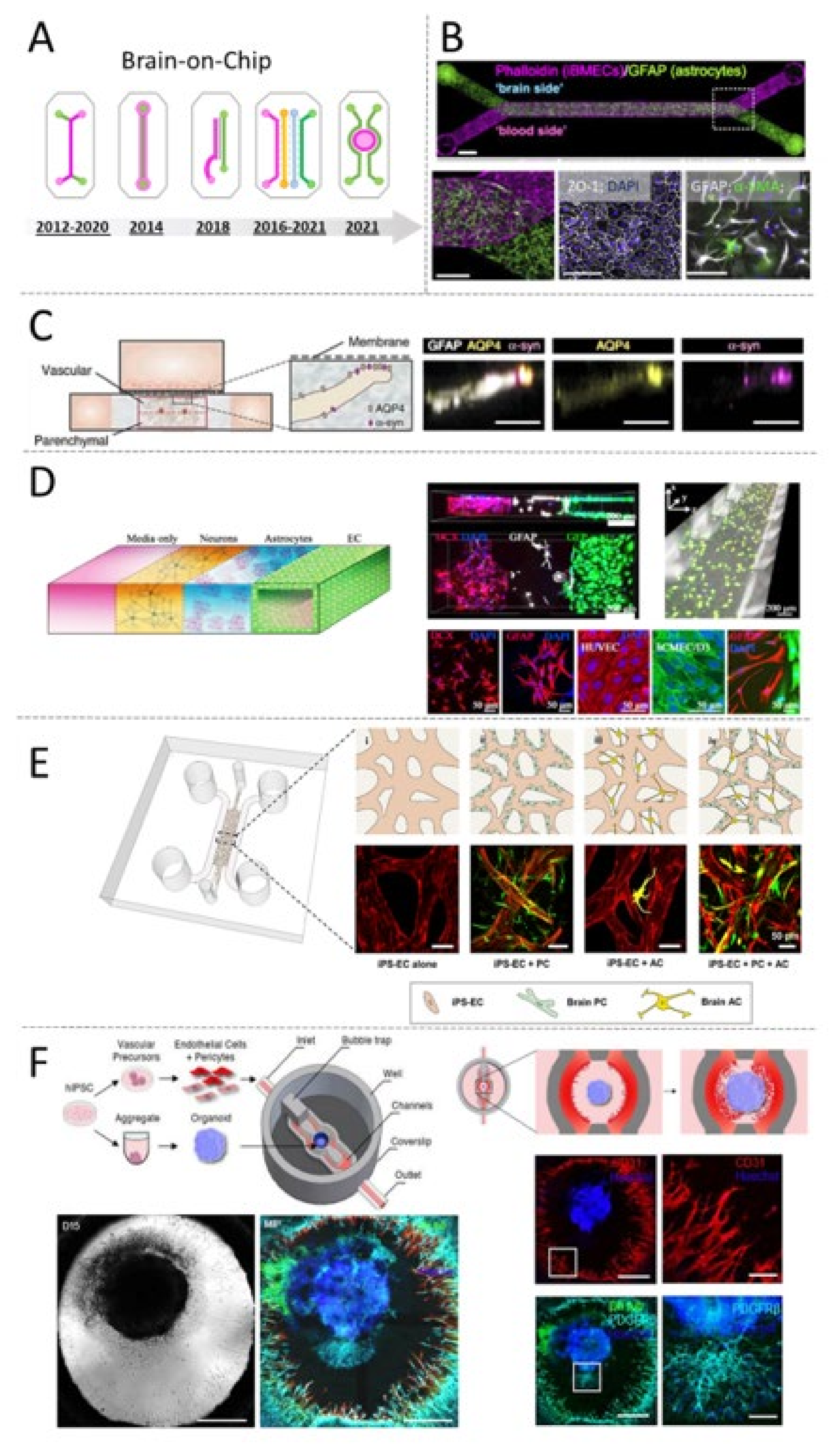
5.2. Tracing the Design Evolution of the Blood-Brain Barrier-on-Chip
5.3. Endothelialized Sandwich Chips

5.4. Chips Incorporating Hydrogels
5.5. Micro-Vascularized Organ-on-Chips
6. The Emergence of Bioprinted Microphysiological Systems


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bioprinting Technique | Biomaterials | Cell Type | Crosslinking | Rheological Properties | Printing Conditions | Ref. |
|---|---|---|---|---|---|---|
| Extrusion | GelMA PDMS | Liver HepG2/C3A | Photo Crosslinking (UV, 850 mW, 15s from 8.5 cm distance) | Elastic module of 5 kPa | Speed: 2 mm/s Pressure: not specified Temp: RT | [122] |
| Extrusion | GelMA | Human adipose tissue-derived mesenchymal stem cells (hAD-MSCs) | Photo Crosslinking (UV, 335 nm) | Shear rate of 0.01 to 1000 s−1 at 25 °C | Speed: 260 mm/min Pressure: 2.8 and 3.8 psi Temp: 30 °C or 37 °C | [123] |
| Coaxial Extrusion | GelMA with gold nanorods and alginate | Cardiac fibroblasts, cardiomyocytes | Photo Crosslinking (UV, 800 mW) | Elastic module of 4.2 ± 0.3 kPa (0.1 mg) and 4.7 ± 0.3 kPa (0.25 mg) | Speed: 1–6 mm/s Pressure: not specified Temp: RT | [124] |
| Extrusion | GelMA, Pluronic PDMS | Human colorectal cancer cells | Photocrosslinking (UV, 400 nm) | Not reported | Speed: 100 mm/min Pressure: 2–4 kPa Temp: 26 °C | [125] |
| Extrusion | Gelatin(G) GelMA(GM) Acetylated GelMA (GMA) | Human adipose-derived stem cells (ASCs), human dermal microvascular endothelial cells (HDMECs) | Photo crosslinking (UV, 365 nm, 0.54 J cm −2, 60 sec) | Storage Modulus: (G) 556.20 Pa (GM) 1373.21 Pa (GMA) 500.25 Pa | Speed: 0.22–0.3 μL/s Pressure: not specified Temp: 21–22 °C | [120] |
| Stereolithography (SLA) | PEGDA GelMA | Primary human mesenchymal cells, Red blood cells (RBCs), HUVECs, hepatocytes | Photocrosslinking (light, 405 nm, 16.4 mW/cm2, 6–40 s to 120 s) | Elastic modulus of PEGDA::10 % strain, 24 kPa | Speed: ≤12ml/hr Resolution: voxels of 250 pL Temp: RT | [121] |
| Stereolithography (SLA) | GelMA with PEGDA | Human bone marrow mesenchymal cells | Photocrosslinking (UV, 355 nm, 45 s) | Elastic modulus: 40% strain | Speed: 2 mm/s Temp: RT | [126] |
| Stereolithography (SLA) | GelMA PEG-bis-(acryloyloxy acetate) | Immortalized hepatocytes (HepaRG) and human stellate cells | Photocrosslinking(blue light illuminator for 30 s/layer) | Not reported | Not specified | [127] |
| Bioprinting Technique | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Inkjet bioprinting | High throughput, speed, precision, reproducibility, wide availability | Thermal and mechanical stresses by Thermal droplet formation, possibility of cell membrane damage due to working frequencies of 15–25kHz in acoustic droplet formation. | [117] |
| Extrusion bioprinting | Broad range of material, economic, universality, versatile, allow customization | Resolution limited to 100μm, only 40% to 80% cell viability, unsuitable for complicated network structure | [116], [128], [129] |
| Stereolithography (SLA) | Higher resolution, cell viability up to 90%, suitable for multiscale network cannels | Expensive, limited number of biocompatible resins, chances of higher level of cytotoxicity | [116], [130], [117] |
| Digital Light Processing (DLP) | Constant high printing speed regardless of complexity of structure, excellent mechanical property and structural integrity, printing accuracy | Expensive resins material; Low mechanical property: risk of crack or deteriorating over time | [131], [132] |
| Laser assisted bioprinting (LAB) | High resolution (varies from pico- to micro-scale, printing speed, high cell activity, precision | Costly due to use of laser source | [116], [133] |
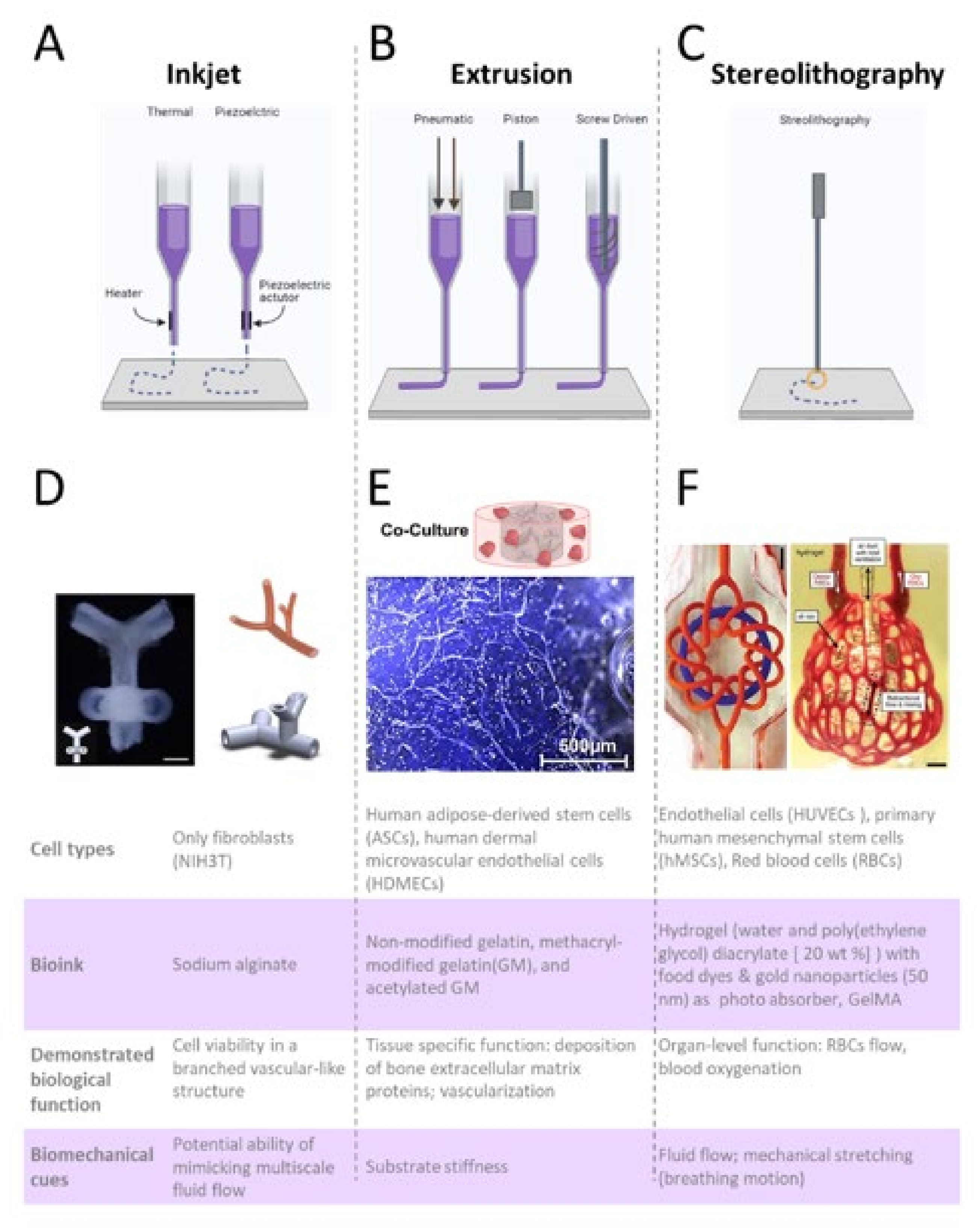
6.1. Extrusion Bioprinting
6.2. Limitations of Extrusion Bioprinting
6.3. Examples of MPSs Obtained via Extrusion Bioprinting
6.4. Stereolithography
6.5. Limitations of Stereolithography
6.6. Examples of MPS Obtained via Stereolithography
6.7. Advantages of Bioprinted MPSs
7. Discussion and Future Directions
Bioprinted MPSs Represent a Fresh Perspective on Strategies to Develop Personalized Models of Human Diseases
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kola, I.; Landis, J. Can the Pharmaceutical Industry Reduce Attrition Rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Pammolli, F.; Righetto, L.; Abrignani, S.; Pani, L.; Pelicci, P.G.; Rabosio, E. The Endless Frontier? The Recent Upsurge of R&D Productivity in Pharmaceuticals. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The Productivity Crisis in Pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Bracken, M.B. Why Animal Studies Are Often Poor Predictors of Human Reactions to Exposure. J. R. Soc. Med. 2009, 102, 120–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.N.; Ingber, D.E. Microfluidic Organs-on-Chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Developmentally Inspired Human ‘Organs on Chips’. Development 2018, 145, dev156125. [Google Scholar] [CrossRef] [Green Version]
- Ergir, E.; Bachmann, B.; Redl, H.; Forte, G.; Ertl, P. Small Force, Big Impact: Next Generation Organ-on-a-Chip Systems Incorporating Biomechanical Cues. Front. Physiol. 2018, 9, 1417. [Google Scholar] [CrossRef]
- Puryear, J.R., III; Yoon, J.-K.; Kim, Y. Advanced Fabrication Techniques of Microengineered Physiological Systems. Micromachines 2020, 11, 730. [Google Scholar] [CrossRef]
- Doke, S.K.; Dhawale, S.C. Alternatives to Animal Testing: A Review. Saudi Pharm. J. 2015, 23, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Carrel, A.; Ebeling, A.H. Action of serum on fibroblasts in vitro. J. Exp. Med. 1923, 37, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H. Nutrition Needs of Mammalian Cells in Tissue Culture. Science 1955, 122, 501–504. [Google Scholar] [CrossRef]
- Alhaque, S.; Themis, M.; Rashidi, H. Three-Dimensional Cell Culture: From Evolution to Revolution. Philos. Trans. Soc. Biol. Sci. 2018, 373, 20170216. [Google Scholar] [CrossRef]
- Holland, I.; Davies, J.A. Automation in the Life Science Research Laboratory. Front. Bioeng. Biotechnol. 2020, 8, 571777. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The Third Dimension Bridges the Gap between Cell Culture and Live Tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Cultures—A Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-Dimensional Cell Culture: The Missing Link in Drug Discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Chen, C.S. Deconstructing the Third Dimension—How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. Adv. Sci. Drug Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A.; Kruuv, J. A Multi-Component Radiation Survival Curve Using an in Vitro Tumour Model. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1970, 18, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Folkman, J. Tumor Angiogensis: Role in Regulation of Tumor Growth. Macromol. Regul. Growth Dev. 1974, 30, 43–52. [Google Scholar]
- Brem, S.; Cotran, R.; Folkman, J. Tumor Angiogenesis: A Quantitative Method for Histologic Grading. J. Natl. Cancer Inst. 1972, 48, 347–356. [Google Scholar]
- Folkman, J.; Hochberg, M. Self-regulatin of growth in three dimensions. J. Exp. Med. 1973, 138, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Simian, M.; Bissell, M.J. Organoids: A Historical Perspective of Thinking in Three Dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Swarm, R.L. Transplantation of a Murine Chondrosarcoma in Mice of Different Inbred Strains. J. Natl. Cancer Inst. 1963. [Google Scholar] [CrossRef]
- Orkin, R.W.; Gehron, P.; McGoodwin, E.B.; Martin, G.R.; Valentine, T.; Swarm, R. A Murine Tumor Producing a Matrix of Basement Membrane. J. Exp. Med. 1977, 145, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, Y.; Qin, S.; Zhao, W.; Chu, Q.; Wu, K. Organoid Technology in Disease Modelling, Drug Development, Personalized Treatment and Regeneration Medicine. Exp. Hematol. Oncol. 2018, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Bartfeld, S.; Clevers, H. Stem Cell-Derived Organoids and Their Application for Medical Research and Patient Treatment. J. Mol. Med. 2017, 95, 729–738. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; de Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; van de Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing Responses to CFTR-Modulating Drugs Using Rectal Organoids Derived from Subjects with Cystic Fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef]
- Takahashi, T. Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef]
- Traub, O.; Berk, B.C. Laminar Shear Stress: Mechanisms by Which Endothelial Cells Transduce an Atheroprotective Force. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Yang, F.; Shu, W.; Chen, Z.; Chen, M. Functional Roles of Shear Stress in Vascular Endothelial Cells. Cell. Immunol. Serum Biol. 2016, 3, 4. [Google Scholar] [CrossRef]
- Huang, Q.; Hu, X.; He, W.; Zhao, Y.; Hao, S.; Wu, Q.; Li, S.; Zhang, S.; Shi, M. Fluid Shear Stress and Tumor Metastasis. Am. J. Cancer Res. 2018, 8, 763–777. [Google Scholar] [PubMed]
- Truskey, G.A.; Fernandez, C.E. Tissue-Engineered Blood Vessels as Promising Tools for Testing Drug Toxicity. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, A.; Albers, H.J.; Passier, R.; Mummery, C.L.; van den Berg, A.; Orlova, V.V.; van der Meer, A.D. Advanced in Vitro Models of Vascular Biology: Human Induced Pluripotent Stem Cells and Organ-on-Chip Technology. Adv. Drug Deliv. Rev. 2019, 140, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sokabe, M. Short history on the birth of mechanobiology. Clin. Calc. 2016, 26, 1659–1669. [Google Scholar]
- Eyckmans, J.; Boudou, T.; Yu, X.; Chen, C.S. A Hitchhiker’s Guide to Mechanobiology. Dev. Cell 2011, 21, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dianzani, M.U. Bizzozero and the Discovery of Platelets. Am. J. Nephrol. 1994, 14, 330–336. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, G.; Cerletti, C. Platelet Adhesion and Aggregation and Fibrin Formation in Flowing Blood: A Historical Contribution by Giulio Bizzozero. Platelets 2002, 13, 85–89. [Google Scholar] [CrossRef]
- Baumgartner, H.R. Effects of Anticoagulation on the Interaction of Human Platelets with Subendothelium in Flowing Blood. Schweiz. Med. Wochenschr. 1976, 106, 1367–1368. [Google Scholar] [PubMed]
- Sakariassen, K.S.; Turitto, V.T.; Baumgartner, H.R. Recollections of the Development of Flow Devices for Studying Mechanisms of Hemostasis and Thrombosis in Flowing Whole Blood. J. Thromb. Haemost. 2004, 2, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, V.; Grabowski, E.; Bini, A.; Nemerson, Y. Platelets, Circulating Tissue Factor, and Fibrin Colocalize in Ex Vivo Thrombi: Real-Time Fluorescence Images of Thrombus Formation and Propagation under Defined Flow Conditions. Blood 2002, 100, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Savage, B.; Sixma, J.J.; Ruggeri, Z.M. Functional Self-Association of von Willebrand Factor during Platelet Adhesion under Flow. Proc. Natl. Acad. Sci. USA 2002, 99, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Neeves, K.B.; Diamond, S.L. A Membrane-Based Microfluidic Device for Controlling the Flux of Platelet Agonists into Flowing Blood. Lab. Chip 2008, 8, 701–709. [Google Scholar] [CrossRef]
- Hosokawa, K.; Ohnishi, T.; Kondo, T.; Fukasawa, M.; Koide, T.; Maruyama, I.; Tanaka, K.A. A Novel Automated Microchip Flow-Chamber System to Quantitatively Evaluate Thrombus Formation and Antithrombotic Agents under Blood Flow Conditions. J. Thromb. Haemost. 2011, 9, 2029–2037. [Google Scholar] [CrossRef]
- Wolff, J. Das Gesetz Der Transformation Der Knochen. Hirshwald 1892, 1, 1–152. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, G.G. Turbulence in Human Intracranial Saccular Aneurysms. J. Neurosurg. 1970, 33, 485–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ali, J.; Sorger, P.K.; Jensen, K.F. Cells on Chips. Nature 2006, 442, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Dewey, C.F.; Bussolari, S.R.; Gordon, E.J.; Gimbrone, M.A. Influence of Hemodynamic Forces on Vascular Endothelial Function. In Vitro Studies of Shear Stress and Pinocytosis in Bovine Aortic Cells. J. Clin. Investig. 1984, 73, 1121–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudenschild, C.C.; Grunwald, J.; Chobanian, A.V. Effects of Hypertension on Migration and Proliferation of Smooth Muscle in Culture. Hypertension 1985, 7, I101. [Google Scholar] [CrossRef] [Green Version]
- Sumpio, B.E.; Banes, A.J.; Levin, L.G.; Johnson, G. Mechanical Stress Stimulates Aortic Endothelial Cells to Proliferate. J. Vasc. Surg. 1987, 6, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Yamawaki, H.; Pan, S.; Lee, R.T.; Berk, B.C. Fluid Shear Stress Inhibits Vascular Inflammation by Decreasing Thioredoxin-Interacting Protein in Endothelial Cells. J. Clin. Investig. 2005, 115, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.A.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(Dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Büttgenbach, S. Electromagnetic Micromotors—Design, Fabrication and Applications. Micromachines 2014, 5, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.; Mai, Q.; Sudhir, K.; Weiss, R.H.; Ives, H.E. Mechanical Strain Induces Growth of Vascular Smooth Muscle Cells via Autocrine Action of PDGF. J. Cell Biol. 1993, 123, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacolley, P. Mechanical Influence of Cyclic Stretch on Vascular Endothelial Cells. Cardiovasc. Res. 2004, 63, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Sin, A.; Chin, K.C.; Jamil, M.F.; Kostov, Y.; Rao, G.; Shuler, M.L. The Design and Fabrication of Three-Chamber Microscale Cell Culture Analog Devices with Integrated Dissolved Oxygen Sensors. Biotechnol. Prog. 2008, 20, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Huh, D.; Fujioka, H.; Tung, Y.-C.; Futai, N.; Paine, R.; Grotberg, J.B.; Takayama, S. Acoustically Detectable Cellular-Level Lung Injury Induced by Fluid Mechanical Stresses in Microfluidic Airway Systems. Proc. Natl. Acad. Sci. USA 2007, 104, 18886–18891. [Google Scholar] [CrossRef] [Green Version]
- Huh, D. A Human Breathing Lung-on-a-Chip. Ann. Am. Thorac. Soc. 2015, 12, S42–S44. [Google Scholar] [CrossRef]
- Beaurivage, C.; Kanapeckaite, A.; Loomans, C.; Erdmann, K.S.; Stallen, J.; Janssen, R.A.J. Development of a Human Primary Gut-on-a-Chip to Model Inflammatory Processes. Sci. Rep. 2020, 10, 21475. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-J.; Otieno, M.A.; Ronxhi, J.; Lim, H.-K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing Human and Cross-Species Drug Toxicities Using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, M.; Palma do Carmo, M.A.; van der Helm, M.W.; Le-The, H.; de Graaf, M.N.S.; Orlova, V.; van den Berg, A.; van der Meer, A.D.; Broersen, K.; Segerink, L.I. Multiplexed Blood—Brain Barrier Organ-on-Chip. Lab. Chip 2020, 20, 3132–3143. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.L.; Fabre, K.M.; Tagle, D.A. The National Institutes of Health Microphysiological Systems Program Focuses on a Critical Challenge in the Drug Discovery Pipeline. Stem Cell Res. Ther. 2013, 4, scrt361. [Google Scholar] [CrossRef] [Green Version]
- Hargrove-Grimes, P.; Low, L.A.; Tagle, D.A. Microphysiological Systems: What It Takes for Community Adoption. Exp. Biol. Med. 2021, 246, 1435–1446. [Google Scholar] [CrossRef]
- Bauer, S.; Wennberg Huldt, C.; Kanebratt, K.P.; Durieux, I.; Gunne, D.; Andersson, S.; Ewart, L.; Haynes, W.G.; Maschmeyer, I.; Winter, A.; et al. Functional Coupling of Human Pancreatic Islets and Liver Spheroids On-a-Chip: Towards a Novel Human Ex Vivo Type 2 Diabetes Model. Sci. Rep. 2017, 7, 14620. [Google Scholar] [CrossRef]
- Eckstrum, K.; Striz, A.; Ferguson, M.; Zhao, Y.; Welch, B.; Solomotis, N.; Olejnik, N.; Sprando, R. Utilization of a Model Hepatotoxic Compound, Diglycolic Acid, to Evaluate Liver Organ-Chip Performance and in Vitro to in Vivo Concordance. Food Chem. Toxicol. 2020, 146, 111850. [Google Scholar] [CrossRef]
- Vriend, J.; Vormann, M.K.; Lanz, H.L.; Joore, J.; Trietsch, S.J.; Russel, F.G.M.; Jacobsen, B.; Roth, A.; Lu, S.; Polli, J.W.; et al. Nephroscreen: A Robust and Versatile Renal Tubule-on-a-Chip Platform for Nephrotoxicity Assessment. Curr. Opin. Toxicol. 2021, 25, 42–48. [Google Scholar] [CrossRef]
- Guo, X.; Smith, V.; Jackson, M.; Tran, M.; Thomas, M.; Patel, A.; Lorusso, E.; Nimbalkar, S.; Cai, Y.; McAleer, C.W.; et al. A Human-Based Functional NMJ System for Personalized ALS Modeling and Drug Testing. Adv. Ther. 2020, 3, 2000133. [Google Scholar] [CrossRef] [PubMed]
- McAleer, C.W.; Long, C.J.; Elbrecht, D.; Sasserath, T.; Bridges, L.R.; Rumsey, J.W.; Martin, C.; Schnepper, M.; Wang, Y.; Schuler, F.; et al. Multi-Organ System for the Evaluation of Efficacy and off-Target Toxicity of Anticancer Therapeutics. Sci. Transl. Med. 2019, 11, eaav1386. [Google Scholar] [CrossRef] [PubMed]
- Roth, A. MPS-WS Berlin 2019 Human Microphysiological Systems for Drug Development. Science 2021, 373, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Thiele, J.; Abdelmohsen, L.; Xu, J.; Huck, W.T.S. Biocompatible Macro-Initiators Controlling Radical Retention in Microfluidic on-Chip Photo-Polymerization of Water-in-Oil Emulsions. Chem. Commun. 2014, 50, 112–114. [Google Scholar] [CrossRef]
- Ren, Y.; Ray, S.; Liu, Y. Reconfigurable Acrylic-Tape Hybrid Microfluidics. Sci. Rep. 2019, 9, 4824. [Google Scholar] [CrossRef]
- Vozzi, G.; Flaim, C.; Ahluwalia, A.; Bhatia, S. Fabrication of PLGA Scaffolds Using Soft Lithography and Microsyringe Deposition. Biomaterials 2003, 24, 2533–2540. [Google Scholar] [CrossRef]
- Sahin, O.; Ashokkumar, M.; Ajayan, P.M. Micro-and Nanopatterning of Biomaterial Surfaces. In Fundamental Biomaterials: Metals; Elsevier: Amsterdam, The Netherlands, 2018; pp. 67–78. ISBN 978-0-08-102205-4. [Google Scholar]
- Mannino, R.G.; Pandian, N.K.R.; Jain, A.; Lam, W.A. Engineering “Endothelialized” Microfluidics for Investigating Vascular and Hematologic Processes Using Non-Traditional Fabrication Techniques. Curr. Opin. Biomed. Eng. 2018, 5, 13–20. [Google Scholar] [CrossRef]
- Khademhosseini, A.; Langer, R. Microengineered Hydrogels for Tissue Engineering. Biomaterials 2007, 28, 5087–5092. [Google Scholar] [CrossRef]
- Iliescu, C.; Taylor, H.; Avram, M.; Miao, J.; Franssila, S. A Practical Guide for the Fabrication of Microfluidic Devices Using Glass and Silicon. Biomicrofluidics 2012, 6, 016505. [Google Scholar] [CrossRef] [Green Version]
- Paoli, R.; Di Giuseppe, D.; Badiola-Mateos, M.; Martinelli, E.; Lopez-Martinez, M.J.; Samitier, J. Rapid Manufacturing of Multilayered Microfluidic Devices for Organ on a Chip Applications. Sensors 2021, 21, 1382. [Google Scholar] [CrossRef]
- Konar, D.; Devarasetty, M.; Yildiz, D.V.; Atala, A.; Murphy, S.V. Lung-On-A-Chip Technologies for Disease Modeling and Drug Development: Supplementary Issue: Image and Video Acquisition and Processing for Clinical Applications. Biomed. Eng. Comput. Biol. 2016, 7, BECB.S34252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, J.; Razavi Bazaz, S.; Aboulkheyr Es, H.; Yaghobian Azari, D.; Thierry, B.; Ebrahimi Warkiani, M.; Ghadiri, M. Lung-on-a-Chip: The Future of Respiratory Disease Models and Pharmacological Studies. Crit. Rev. Biotechnol. 2020, 40, 213–230. [Google Scholar] [CrossRef]
- Rhee, S. Fibroblasts in Three Dimensional Matrices: Cell Migration and Matrix Remodeling. Exp. Mol. Med. 2009, 41, 858. [Google Scholar] [CrossRef] [Green Version]
- Humayun, M.; Chow, C.-W.; Young, E.W.K. Microfluidic Lung Airway-on-a-Chip with Arrayable Suspended Gels for Studying Epithelial and Smooth Muscle Cell Interactions. Lab. Chip 2018, 18, 1298–1309. [Google Scholar] [CrossRef]
- Park, J.Y.; Ryu, H.; Lee, B.; Ha, D.-H.; Ahn, M.; Kim, S.; Kim, J.Y.; Jeon, N.L.; Cho, D.-W. Development of a Functional Airway-on-a-Chip by 3D Cell Printing. Biofabrication 2018, 11, 015002. [Google Scholar] [CrossRef]
- Varone, A.; Nguyen, J.K.; Leng, L.; Barrile, R.; Sliz, J.; Lucchesi, C.; Wen, N.; Gravanis, A.; Hamilton, G.A.; Karalis, K.; et al. A Novel Organ-Chip System Emulates Three-Dimensional Architecture of the Human Epithelia and the Mechanical Forces Acting on It. Biomaterials 2021, 275, 120957. [Google Scholar] [CrossRef]
- Zamprogno, P.; Wüthrich, S.; Achenbach, S.; Thoma, G.; Stucki, J.D.; Hobi, N.; Schneider-Daum, N.; Lehr, C.-M.; Huwer, H.; Geiser, T.; et al. Second-Generation Lung-on-a-Chip with an Array of Stretchable Alveoli Made with a Biological Membrane. Commun. Biol. 2021, 4, 168. [Google Scholar] [CrossRef]
- Seo, J.; Byun, W.Y.; Alisafaei, F.; Georgescu, A.; Yi, Y.-S.; Massaro-Giordano, M.; Shenoy, V.B.; Lee, V.; Bunya, V.Y.; Huh, D. Multiscale Reverse Engineering of the Human Ocular Surface. Nat. Med. 2019, 25, 1310–1318. [Google Scholar] [CrossRef]
- Lee, M.; Hwang, J.-H.; Lim, K.-M. Alternatives to In Vivo Draize Rabbit Eye and Skin Irritation Tests with a Focus on 3D Reconstructed Human Cornea-Like Epithelium and Epidermis Models. Toxicol. Res. 2017, 33, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.B.; Yang, A.C.; Yousef, H.; Lee, D.; Chen, W.; Schaum, N.; Lehallier, B.; Quake, S.R.; Wyss-Coray, T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020, 30, 4418–4432.e4. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, J.G.; Jamieson, J.J.; Linville, R.M.; Searson, P.C. Benchmarking In Vitro Tissue-Engineered Blood—Brain Barrier Models. Fluids Barriers CNS 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In Vitro Models of the Blood—Brain Barrier: An Overview of Commonly Used Brain Endothelial Cell Culture Models and Guidelines for Their Use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-Chip: Recent Breakthroughs and Future Prospects. Biomed. Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, O.; Pamies, D.; Lavell, E.; Schiapparelli, P.; Feng, Y.; Hartung, T.; Bal-Price, A.; Hogberg, H.T.; Quinones-Hinojosa, A.; Guerrero-Cazares, H.; et al. Brain-on-a-Chip Model Enables Analysis of Human Neuronal Differentiation and Chemotaxis. Lab. Chip 2016, 16, 4152–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neal, E.H.; Marinelli, N.A.; Shi, Y.; McClatchey, P.M.; Balotin, K.M.; Gullett, D.R.; Hagerla, K.A.; Bowman, A.B.; Ess, K.C.; Wikswo, J.P.; et al. A Simplified, Fully Defined Differentiation Scheme for Producing Blood-Brain Barrier Endothelial Cells from Human IPSCs. Stem Cell Rep. 2019, 12, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Dolmetsch, R.; Geschwind, D.H. The Human Brain in a Dish: The Promise of IPSC-Derived Neurons. Cell 2011, 145, 831–834. [Google Scholar] [CrossRef] [Green Version]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human IPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005.e6. [Google Scholar] [CrossRef] [PubMed]
- Barrile, R.; van der Meer, A.D.; Park, H.; Fraser, J.P.; Simic, D.; Teng, F.; Conegliano, D.; Nguyen, J.; Jain, A.; Zhou, M.; et al. Organ-on-Chip Recapitulates Thrombosis Induced by an Anti-CD154 Monoclonal Antibody: Translational Potential of Advanced Microengineered Systems. Clin. Pharmacol. Ther. 2018, 104, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Miccoli, B.; Braeken, D.; Li, Y.-C.E. Brain-on-a-Chip Devices for Drug Screening and Disease Modeling Applications. Curr. Pharm. Des. 2019, 24, 5419–5436. [Google Scholar] [CrossRef]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-Enhanced Blood-Brain Barrier Chip Recapitulates Human Barrier Function and Shuttling of Drugs and Antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef]
- Lee, C.S.; Leong, K.W. Advances in Microphysiological Blood-Brain Barrier (BBB) Models towards Drug Delivery. Curr. Opin. Biotechnol. 2020, 66, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Tourovskaia, A.; Fauver, M.; Kramer, G.; Simonson, S.; Neumann, T. Tissue-Engineered Microenvironment Systems for Modeling Human Vasculature. Exp. Biol. Med. 2014, 239, 1264–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herland, A.; van der Meer, A.D.; FitzGerald, E.A.; Park, T.-E.; Sleeboom, J.J.F.; Ingber, D.E. Distinct Contributions of Astrocytes and Pericytes to Neuroinflammation Identified in a 3D Human Blood-Brain Barrier on a Chip. PLoS ONE 2016, 11, e0150360. [Google Scholar] [CrossRef] [Green Version]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A Perfused Human Blood—Brain Barrier on-a-Chip for High-Throughput Assessment of Barrier Function and Antibody Transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.I.; Sei, Y.J.; Park, H.-J.; Kim, J.; Ryu, Y.; Choi, J.J.; Sung, H.-J.; MacDonald, T.J.; Levey, A.I.; Kim, Y. Microengineered Human Blood—Brain Barrier Platform for Understanding Nanoparticle Transport Mechanisms. Nat. Commun. 2020, 11, 175. [Google Scholar] [CrossRef]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L.K. A 3D Neurovascular Microfluidic Model Consisting of Neurons, Astrocytes and Cerebral Endothelial Cells as a Blood—Brain Barrier. Lab. Chip 2017, 17, 448–459. [Google Scholar] [CrossRef]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D Self-Organized Microvascular Model of the Human Blood-Brain Barrier with Endothelial Cells, Pericytes and Astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Hajal, C.; Shin, Y.; Li, L.; Serrano, J.C.; Jacks, T.; Kamm, R.D. The CCL2-CCR2 Astrocyte-Cancer Cell Axis in Tumor Extravasation at the Brain. Sci. Adv. 2021, 7, eabg8139. [Google Scholar] [CrossRef]
- Salmon, I.; Grebenyuk, S.; Fattah, A.R.A.; Rustandi, G.; Pilkington, T.; Verfaillie, C.; Ranga, A. Engineering Neurovascular Organoids with 3D Printed Microfluidic Chips. bioRxiv 2021. [Google Scholar] [CrossRef]
- Raimondi, I.; Tunesi, M.; Forloni, G.; Albani, D.; Giordano, C. 3D Brain Tissue Physiological Model with Co-Cultured Primary Neurons and Glial Cells in Hydrogels. J. Tissue Eng. 2020, 11, 204173142096398. [Google Scholar] [CrossRef]
- Hyvärinen, T.; Hagman, S.; Ristola, M.; Sukki, L.; Veijula, K.; Kreutzer, J.; Kallio, P.; Narkilahti, S. Co-Stimulation with IL-1β and TNF-α Induces an Inflammatory Reactive Astrocyte Phenotype with Neurosupportive Characteristics in a Human Pluripotent Stem Cell Model System. Sci. Rep. 2019, 9, 16944. [Google Scholar] [CrossRef]
- Tahergorabi, Z.; Khazaei, M. A Review on Angiogenesis and Its Assays. Iran. J. Basic Med. Sci. 2012, 15, 1110–1126. [Google Scholar] [PubMed]
- Kim, S.; Kim, W.; Lim, S.; Jeon, J. Vasculature-On-A-Chip for In Vitro Disease Models. Bioengineering 2017, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Thayer, P.; Martinez, H.; Gatenholm, E. History and Trends of 3D Bioprinting. In 3D Bioprinting; Crook, J.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2140, pp. 3–18. ISBN 978-1-07-160519-6. [Google Scholar]
- Miri, A.K.; Mostafavi, E.; Khorsandi, D.; Hu, S.-K.; Malpica, M.; Khademhosseini, A. Bioprinters for Organs-on-Chips. Biofabrication 2019, 11, 042002. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jia, Y.; Dong, H.; Dong, D.; Zheng, J. Combining Additive Manufacturing with Microfluidics: An Emerging Method for Developing Novel Organs-on-Chips. Curr. Opin. Chem. Eng. 2020, 28, 1–9. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A Practical Guide to Hydrogels for Cell Culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.; Xu, C.; Chai, W.; Zhang, Z.; Fu, J.; Huang, Y. Freeform Inkjet Printing of Cellular Structures with Bifurcations: Approach Freeform Fabrication of Bifurcated Cellular Structures by Using a Liquid Support-Based Inkjet Printing Approach. Biotechnol. Bioeng. 2015, 112, 1047–1055. [Google Scholar] [CrossRef]
- Leucht, A.; Volz, A.-C.; Rogal, J.; Borchers, K.; Kluger, P.J. Advanced Gelatin-Based Vascularization Bioinks for Extrusion-Based Bioprinting of Vascularized Bone Equivalents. Sci. Rep. 2020, 10, 5330. [Google Scholar] [CrossRef] [Green Version]
- Grigoryan, B.; Paulsen, S.J.; Corbett, D.C.; Sazer, D.W.; Fortin, C.L.; Zaita, A.J.; Greenfield, P.T.; Calafat, N.J.; Gounley, J.P.; Ta, A.H.; et al. Multivascular Networks and Functional Intravascular Topologies within Biocompatible Hydrogels. Science 2019, 364, 458–464. [Google Scholar] [CrossRef]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Shrike Zhang, Y.; Shin, S.R.; Calzone, G.; et al. A Liver-on-a-Chip Platform with Bioprinted Hepatic Spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef]
- Pepelanova, I.; Kruppa, K.; Scheper, T.; Lavrentieva, A. Gelatin-Methacryloyl (GelMA) Hydrogels with Defined Degree of Functionalization as a Versatile Toolkit for 3D Cell Culture and Extrusion Bioprinting. Bioengineering 2018, 5, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Shin, S.R.; van Kempen, T.; Li, Y.; Ponraj, V.; Nasajpour, A.; Mandla, S.; Hu, N.; Liu, X.; Leijten, J.; et al. Gold Nanocomposite Bioink for Printing 3D Cardiac Constructs. Adv. Funct. Mater. 2017, 27, 1605352. [Google Scholar] [CrossRef] [PubMed]
- Mazrouei, R.; Velasco, V.; Esfandyarpour, R. 3D-Bioprinted All-Inclusive Bioanalytical Platforms for Cell Studies. Sci. Rep. 2020, 10, 14669. [Google Scholar] [CrossRef]
- Zhu, W.; Cui, H.; Boualam, B.; Masood, F.; Flynn, E.; Rao, R.D.; Zhang, Z.-Y.; Zhang, L.G. 3D Bioprinting Mesenchymal Stem Cell-Laden Construct with Core–Shell Nanospheres for Cartilage Tissue Engineering. Nanotechnology 2018, 29, 185101. [Google Scholar] [CrossRef]
- Grix, T.; Ruppelt, A.; Thomas, A.; Amler, A.-K.; Noichl, B.; Lauster, R.; Kloke, L. Bioprinting Perfusion-Enabled Liver Equivalents for Advanced Organ-on-a-Chip Applications. Genes 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Grover, H.; Spatarelu, C.-P.; De’De’, K.; Zhao, S.; Yang, K.; Shrike Zhang, Y.; Chen, Z. 1 Thayer School of Engineering, Dartmouth College, Hanover, NH 03755, USA; 2 Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, MA 02139 Vascularization in 3D Printed Tissues: Emerging Technologies to Overcome Longstanding Obstacles. AIMS Cell Tissue Eng. 2018, 2, 163–184. [Google Scholar] [CrossRef]
- Zorlutuna, P.; Annabi, N.; Camci-Unal, G.; Nikkhah, M.; Cha, J.M.; Nichol, J.W.; Manbachi, A.; Bae, H.; Chen, S.; Khademhosseini, A. Microfabricated Biomaterials for Engineering 3D Tissues. Adv. Mater. 2012, 24, 1782–1804. [Google Scholar] [CrossRef] [Green Version]
- Rana Khalid, I.; Darakhshanda, I.; Rafia, R. 3D Bioprinting: An Attractive Alternative to Traditional Organ Transplantation. Arch. Biomed. Sci. Eng. 2019, 5, 007–018. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Fu, J.; Lin, H.; He, Y. Development of 3D Bioprinting: From Printing Methods to Biomedical Applications. Asian J. Pharm. Sci. 2020, 15, 529–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, Q.; Wang, S.; Tao, J.; Gou, M. Digital Light Processing Based Three-Dimensional Printing for Medical Applications. Int. J. Bioprint. 2019, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, M.; Fan, X.; Zhou, H. Recent Advances in Bioprinting Techniques: Approaches, Applications and Future Prospects. J. Transl. Med. 2016, 14, 271. [Google Scholar] [CrossRef] [Green Version]
- Chopin-Doroteo, M.; Mandujano-Tinoco, E.A.; Krötzsch, E. Tailoring of the Rheological Properties of Bioinks to Improve Bioprinting and Bioassembly for Tissue Replacement. Biochim. Biophys. Acta BBA Gen. Subj. 2021, 1865, 129782. [Google Scholar] [CrossRef]
- Schwab, A.; Levato, R.; D’Este, M.; Piluso, S.; Eglin, D.; Malda, J. Printability and Shape Fidelity of Bioinks in 3D Bioprinting. Chem. Rev. 2020, 120, 11028–11055. [Google Scholar] [CrossRef]
- Zur Nieden, N.I.; Turgman, C.C.; Lang, X.; Larsen, J.M.; Granelli, J.; Hwang, Y.-J.; Lyubovitsky, J.G. Fluorescent Hydrogels for Embryoid Body Formation and Osteogenic Differentiation of Embryonic Stem Cells. ACS Appl. Mater. Interfaces 2015, 7, 10599–10605. [Google Scholar] [CrossRef]
- Mancha Sánchez, E.; Gómez-Blanco, J.C.; López Nieto, E.; Casado, J.G.; Macías-García, A.; Díaz Díez, M.A.; Carrasco-Amador, J.P.; Torrejón Martín, D.; Sánchez-Margallo, F.M.; Pagador, J.B. Hydrogels for Bioprinting: A Systematic Review of Hydrogels Synthesis, Bioprinting Parameters, and Bioprinted Structures Behavior. Front. Bioeng. Biotechnol. 2020, 8, 776. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Barua, R.; Das, J. Importance of Alginate Bioink for 3D Bioprinting in Tissue Engineering and Regenerative Medicine. In Alginates Recent Uses of This Natural Polymer; Pereira, L., Ed.; IntechOpen: London, UK, 2020; ISBN 978-1-78985-641-5. [Google Scholar]
- Xiao, S.; Zhao, T.; Wang, J.; Wang, C.; Du, J.; Ying, L.; Lin, J.; Zhang, C.; Hu, W.; Wang, L.; et al. Gelatin Methacrylate (GelMA)-Based Hydrogels for Cell Transplantation: An Effective Strategy for Tissue Engineering. Stem Cell Rev. Rep. 2019, 15, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Kamdem Tamo, A.; Doench, I.; Walter, L.; Montembault, A.; Sudre, G.; David, L.; Morales-Helguera, A.; Selig, M.; Rolauffs, B.; Bernstein, A.; et al. Development of Bioinspired Functional Chitosan/Cellulose Nanofiber 3D Hydrogel Constructs by 3D Printing for Application in the Engineering of Mechanically Demanding Tissues. Polymers 2021, 13, 1663. [Google Scholar] [CrossRef]
- Yue, K.; Trujillo-de Santiago, G.; Alvarez, M.M.; Tamayol, A.; Annabi, N.; Khademhosseini, A. Synthesis, Properties, and Biomedical Applications of Gelatin Methacryloyl (GelMA) Hydrogels. Biomaterials 2015, 73, 254–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-Laden Microengineered Gelatin Methacrylate Hydrogels. Biomaterials 2010, 31, 5536–5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, A.; Clegg, J.R.; Anselmo, A.C.; Mitragotri, S. Hydrogels in the Clinic. Bioeng. Transl. Med. 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesky, D.B.; Truby, R.L.; Gladman, A.S.; Busbee, T.A.; Homan, K.A.; Lewis, J.A. 3D Bioprinting of Vascularized, Heterogeneous Cell-Laden Tissue Constructs. Adv. Mater. 2014, 26, 3124–3130. [Google Scholar] [CrossRef]
- Miri, A.K.; Khalilpour, A.; Cecen, B.; Maharjan, S.; Shin, S.R.; Khademhosseini, A. Multiscale Bioprinting of Vascularized Models. Biomaterials 2019, 198, 204–216. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.-J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A Bioprinted Human-Glioblastoma-on-a-Chip for the Identification of Patient-Specific Responses to Chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Bova, L.; Billi, F.; Cimetta, E. Mini-Review: Advances in 3D Bioprinting of Vascularized Constructs. Biol. Direct 2020, 15, 22. [Google Scholar] [CrossRef]
- Park, J.H.; Jang, J.; Lee, J.-S.; Cho, D.-W. Three-Dimensional Printing of Tissue/Organ Analogues Containing Living Cells. Ann. Biomed. Eng. 2017, 45, 180–194. [Google Scholar] [CrossRef]
- Chia, H.N.; Wu, B.M. Recent Advances in 3D Printing of Biomaterials. J. Biol. Eng. 2015, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Mooney, D.J. Designing Hydrogels for Controlled Drug Delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]




| Fabrication Method | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Soft Lithography | Time saving, inexpensive, reusable molds from single master, easier setup, high throughput, wide range of resolution nano to micrometer | Structure needs to be continuous, other technique such as photolithography, e beam is required to fabricate master stamp once | [76,77] |
| Photolithography | Robust, repeatable, resolution ranges from micron to millimeter | Requires expensive equipment, and specialized training, time consuming, limited to photocrosslinkable materials | [78,79] |
| Etching | High resolution, sub micrometer size, well known technique | Chemicals(acid/base) may damage the polymers, Poor process control and selectivity due to temperature gradient, multiple steps, high cost and time | [9,80] |
| Laser cutting | Robust, time saving, allows for alternative plastics that replace PDMS | Laser cutting of plastic components can be expensive as it requires the use of ventilated rooms or instruments designed to remove harmful gas. | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pun, S.; Haney, L.C.; Barrile, R. Modelling Human Physiology on-Chip: Historical Perspectives and Future Directions. Micromachines 2021, 12, 1250. https://doi.org/10.3390/mi12101250
Pun S, Haney LC, Barrile R. Modelling Human Physiology on-Chip: Historical Perspectives and Future Directions. Micromachines. 2021; 12(10):1250. https://doi.org/10.3390/mi12101250
Chicago/Turabian StylePun, Sirjana, Li Cai Haney, and Riccardo Barrile. 2021. "Modelling Human Physiology on-Chip: Historical Perspectives and Future Directions" Micromachines 12, no. 10: 1250. https://doi.org/10.3390/mi12101250