Electrochemical Evaluation of a Multi-Site Clinical Depth Recording Electrode for Monitoring Cerebral Tissue Oxygen

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Characterization of the Electrode Surface—Morphology and Chemical Analysis
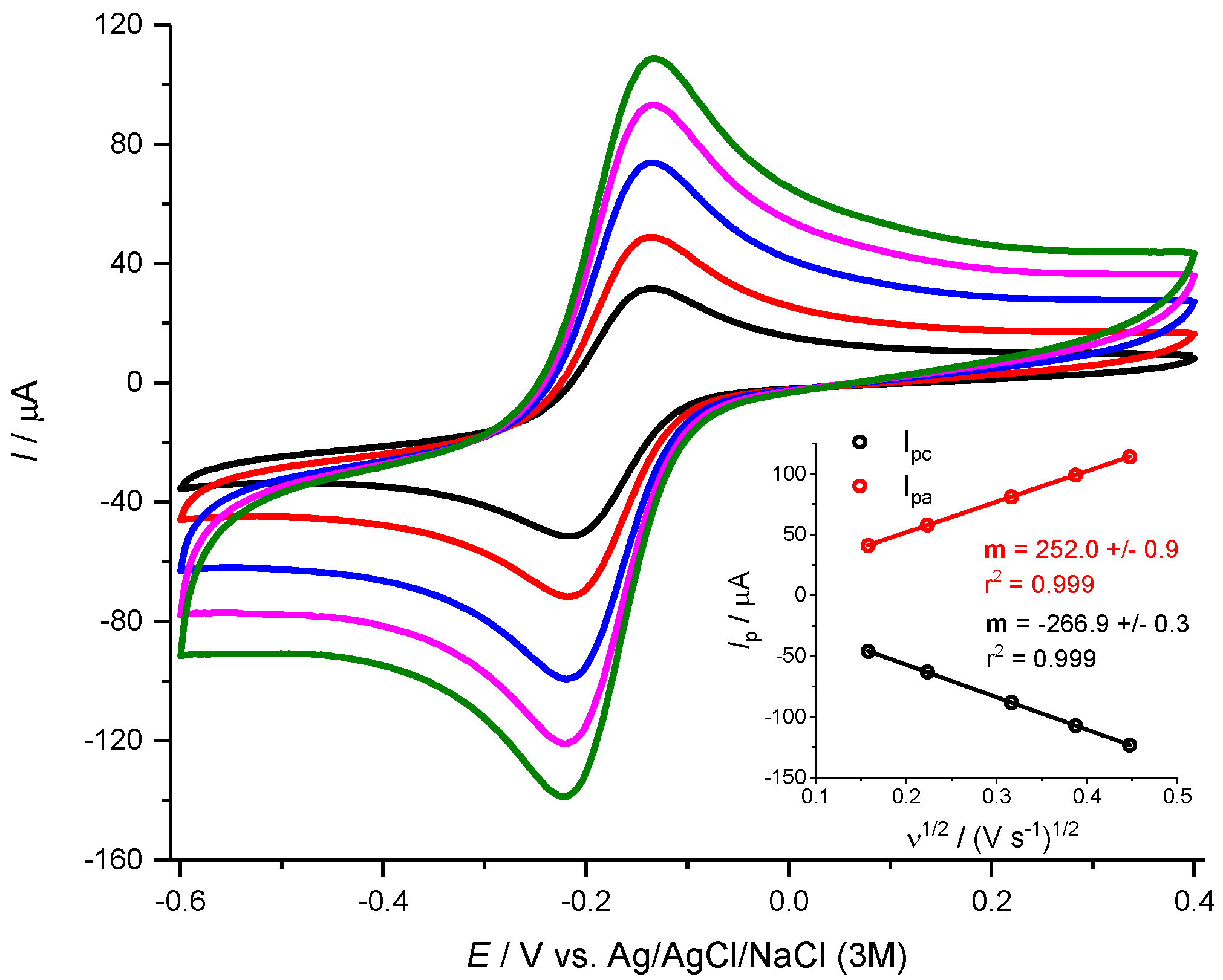
3.2. Electrochemical Active Surface Area
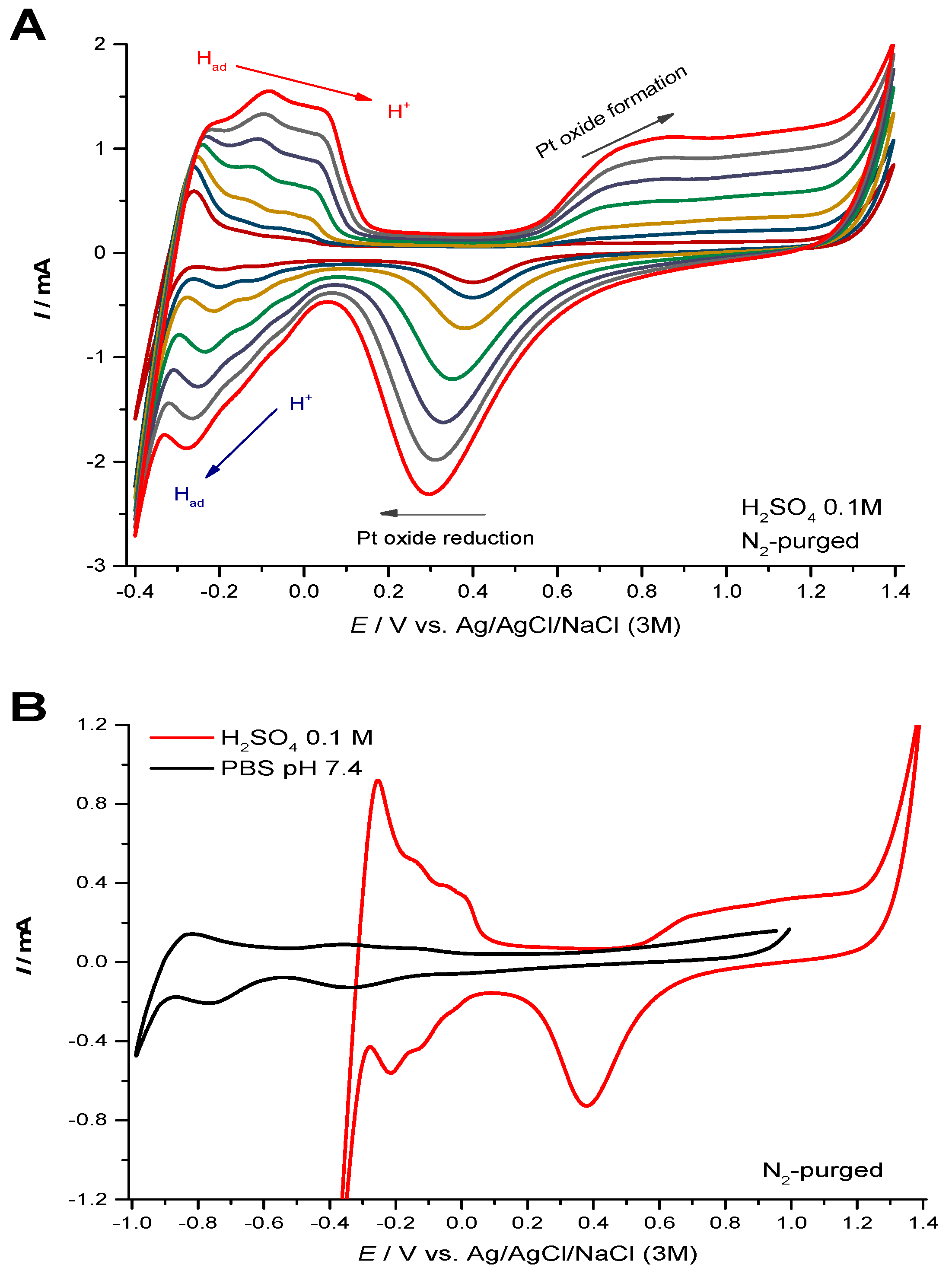
3.3. Electrochemical Behavior in Acidic Electrolyte and in Neutral PBS
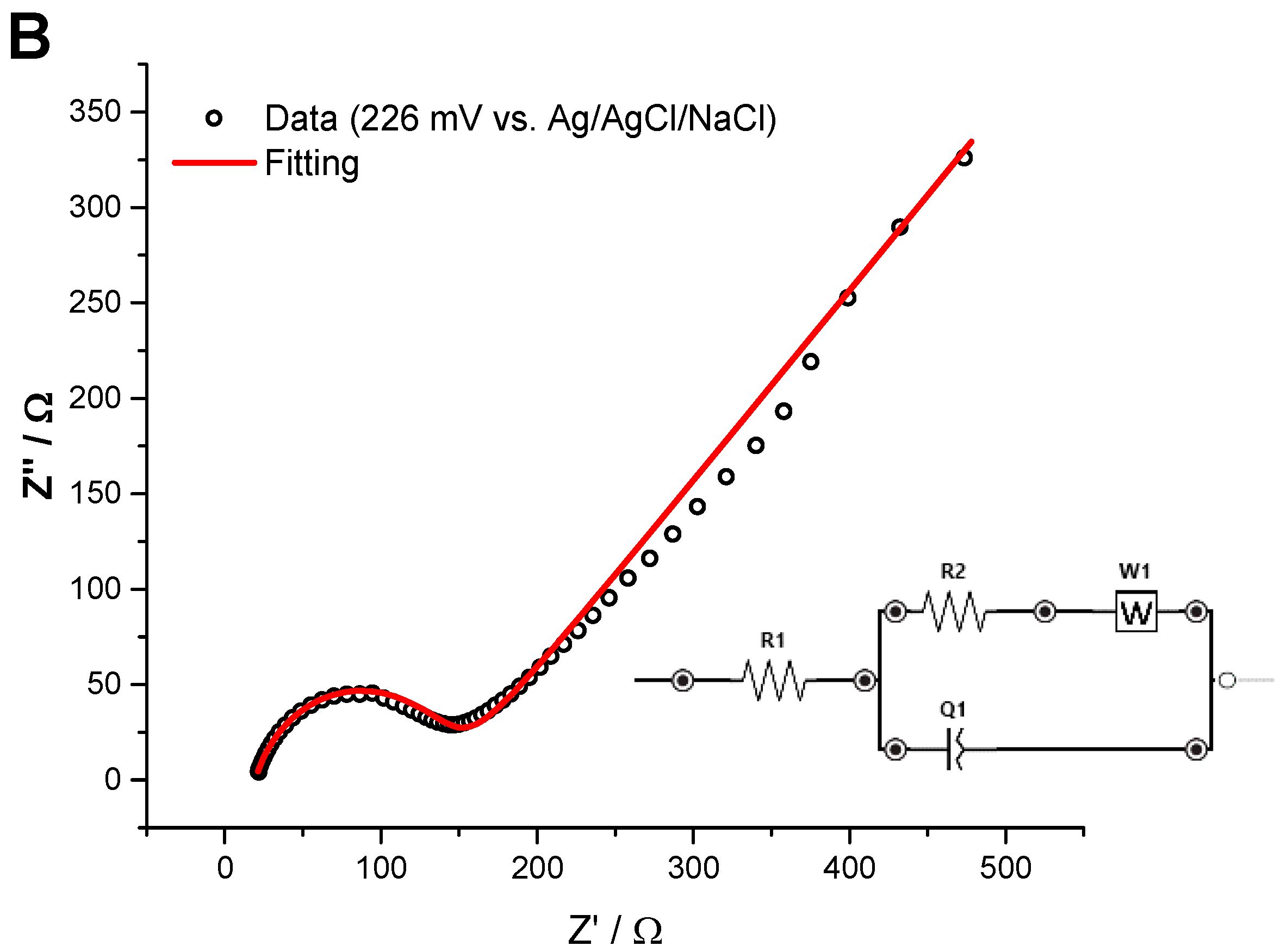
3.4. Electrochemical Impedance Spectroscopy
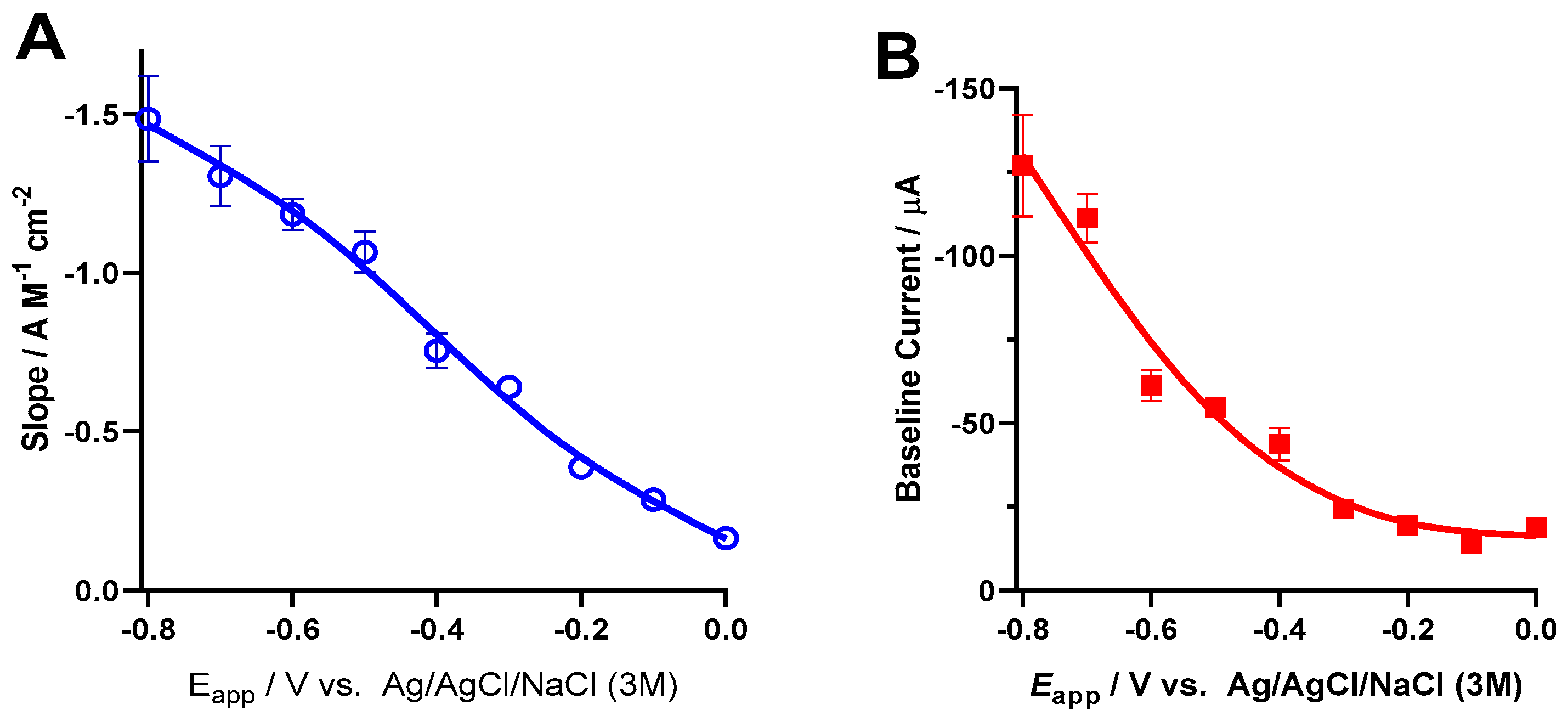
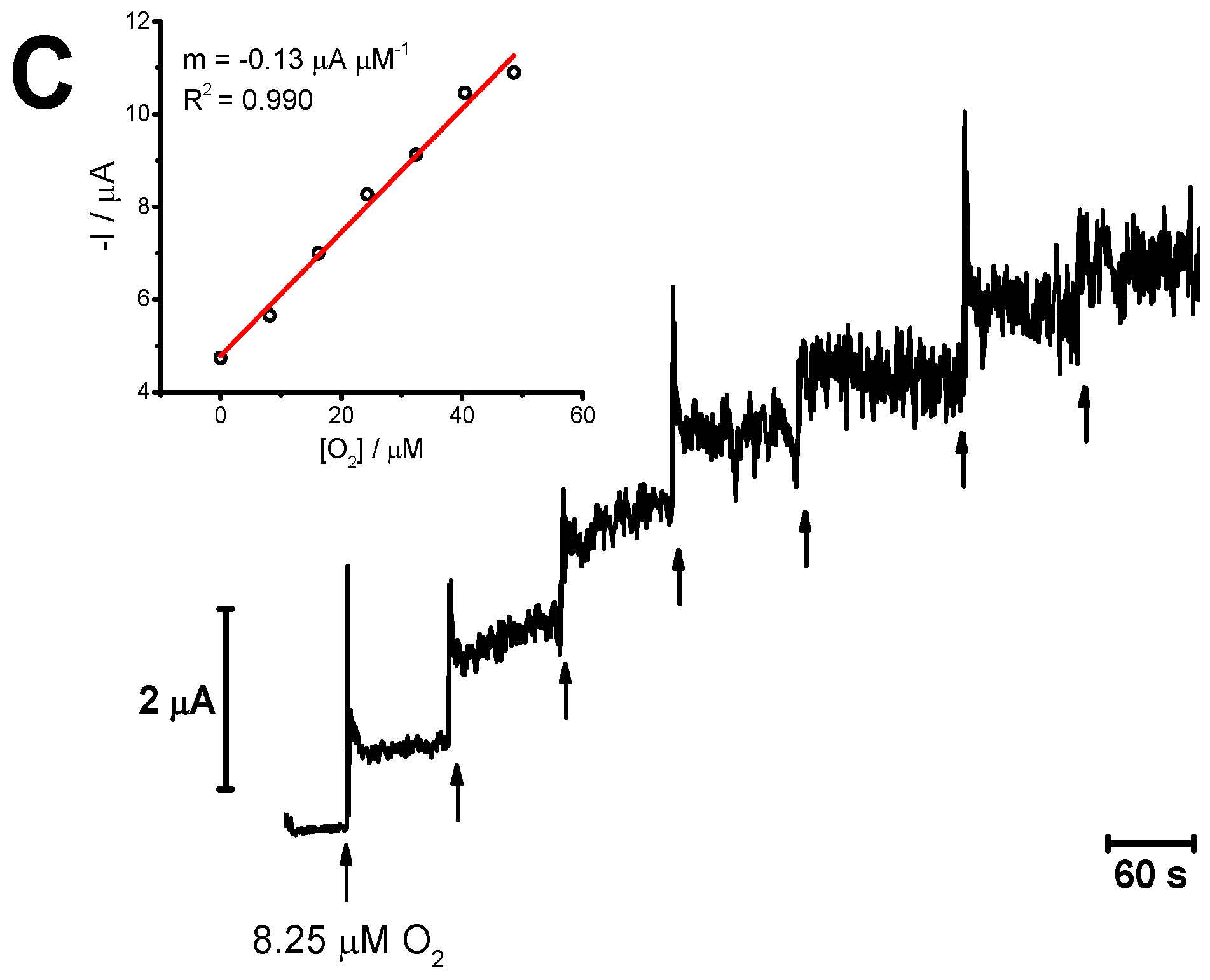
3.5. Oxygen Reduction Reaction at the Platinum Surface
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tasneem, N.; Samaniego, E.A.; Pieper, C.; Leira, E.C.; Adams, H.P.; Hasan, D.; Ortega-Gutierrez, S. Brain Multimodality Monitoring: A New Tool in Neurocritical Care of Comatose Patients. Crit. Care Res. Pract. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheeren, T.W.L.; Kuizenga, M.H.; Maurer, H.; Struys, M.M.R.F.; Heringlake, M. Electroencephalography and Brain Oxygenation Monitoring in the Perioperative Period. Anesth. Analg. 2019, 128, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.W.; Jaeger, M. Systematic and comprehensive literature review of publications on direct cerebral oxygenation monitoring. Open Crit. Care Med. J. 2013, 6, 1–24. [Google Scholar] [CrossRef]
- Bohman, L.E.; Pisapia, J.M.; Sanborn, M.R.; Frangos, S.; Lin, E.; Kumar, M.; Park, S.; Kofke, W.A.; Stiefel, M.F.; Leroux, P.D.; et al. Response of brain oxygen to therapy correlates with long-term outcome after subarachnoid hemorrhage. Neurocrit. Care 2013, 19, 320–328. [Google Scholar] [CrossRef]
- Stiefel, M.F.; Spiotta, A.; Gracias, V.H.; Garuffe, A.M.; Guillamondegui, O.; Maloney-Wilensky, E.; Bloom, S.; Grady, M.S.; LeRoux, P.D. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen monitoring. J. Neurosurg. 2005, 103, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.I.; Fleckenstein, W.; de Jong, D.A.; van Santbrink, H. Monitoring cerebral oxygenation Experimental studies and preliminary clinical results of continuous monitoring of cerebrospinal fluid and brain tissue oxygen tension. Acta Neurochir. Suppl. (Wien) 1993, 59, 50–57. [Google Scholar] [CrossRef]
- Rosner, M.J.; Zauner, A.; Bullock, M.R.R.; Levy, M.L. Clinical experience with 118 brain tissue oxygen partial pressure catheter probes: Comments. Neurosurgery 1998, 43, 1094–1095. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Fleckenstein, W.; de Jong, D.A.; Wolf, M. Effect of Increased ICP and Decreased Cerebral Perfusion Pressure on Brain Tissue and Cerebrospinal Fluid Oxygen Tension. In Proceedings of the Intracranial Pressure VIII; Avezaat, C.J.J., van Eijndhoven, J.H.M., Maas, A.I.R., Tans, J.T.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 233–237. [Google Scholar]
- Kiening, K.L.; Unterberg, A.W.; Bardt, T.F.; Schneider, G.H.; Lanksch, W.R. Monitoring of cerebral oxygenation in patients with severe head injuries: Brain tissue PO2 versus jugular vein oxygen saturation. J. Neurosurg. 1996, 85, 751–757. [Google Scholar] [CrossRef]
- Bratton, S.L.; Chestnut, R.M.; Ghajar, J.; McConnell Hammond, F.F.; Harris, O.A.; Hartl, R.; Manley, G.T.; Nemecek, A.; Newell, D.W.; Rosenthal, G.; et al. Brain Oxygen Monitoring and Thresholds. J. Neurotrauma 2007, 24, S65–S70. [Google Scholar] [CrossRef]
- Bhatia, A.; Gupta, A.K. Neuromonitoring in the intensive care unit. II. Cerebral oxygenation monitoring and microdialysis. Intensive Care Med. 2007, 33, 1322–1328. [Google Scholar] [CrossRef]
- Salehi, A.; Zhang, J.H.; Obenaus, A. Response of the cerebral vasculature following traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2320–2339. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Kalanuria, A. Cerebral Microdialysis in Neurocritical Care. Curr. Neurol. Neurosci. Rep. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Oddo, M.; Levine, J.M.; MacKenzie, L.; Frangos, S.; Feihl, F.; Kasner, S.E.; Katsnelson, M.; Pukenas, B.; MacMurtrie, E.; Maloney-Wilensky, E.; et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery 2011, 69, 1037–1045. [Google Scholar] [CrossRef]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; William Shuttleworth, C.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef]
- Hartings, J.A. Spreading depolarization monitoring in neurocritical care of acute brain injury. Curr. Opin. Crit. Care 2017, 23, 94–102. [Google Scholar] [CrossRef]
- Metzen, E.; Ratcliffe, P.J. HIF hydroxylation and cellular oxygen sensing. Biol. Chem. 2004, 385, 223–230. [Google Scholar] [CrossRef]
- De Georgia, M.A. Brain Tissue Oxygen Monitoring in Neurocritical Care. J. Intensive Care Med. 2015, 30, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Murkin, J.M.; Arango, M. Near-infrared spectroscopy as an index of brain and tissue oxygenation. Br. J. Anaesth. 2009, 103, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Huschak, G.; Hoell, T.; Hohaus, C.; Kern, C.; Minkus, Y.; Meisel, H.-J. Clinical Evaluation of a New Multiparameter Neuromonitoring Device: Measurement of Brain Tissue Oxygen, Brain Temperature, and Intracranial Pressure. J. Neurosurg. Anesthesiol. 2009, 21, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.; Haitsma, I.; Zador, Z.; Hemphill, J.C.; Morabito, D.; Manley, G.; Rosenthal, G. The new licox combined brain tissue oxygen and brain temperature monitor: Assessment of in vitro accuracy and clinical experience in severe traumatic brain injury. Neurosurgery 2008, 63, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, R.I.; Papkovsky, D.B. Optical probes and techniques for O 2 measurement in live cells and tissue. Cell. Mol. Life Sci. 2012, 69, 2025–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkman, M.A.; Smith, M. Brain Oxygenation Monitoring. Anesthesiol. Clin. 2016, 34, 537–556. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.; Woodward, B. A healthy future: Platinum in medical applications platinum group metals enhance the quality of life of the global population. Platin. Met. Rev. 2011, 55, 98–107. [Google Scholar] [CrossRef]
- Cogan, S.F. Neural Stimulation and Recording Electrodes. Annu. Rev. Biomed. Eng. 2008, 10, 275–309. [Google Scholar] [CrossRef] [Green Version]
- Ledo, A.; Lourenço, C.F.; Laranjinha, J.; Brett, C.M.A.; Gerhardt, G.A.; Barbosa, R.M. Ceramic-Based Multisite Platinum Microelectrode Arrays: Morphological Characteristics and Electrochemical Performance for Extracellular Oxygen Measurements in Brain Tissue. Anal. Chem. 2017, 89, 1674–1683. [Google Scholar] [CrossRef]
- Wu, J.; Yang, H. Platinum-based oxygen reduction electrocatalysts. Acc. Chem. Res. 2013, 46, 1848–1857. [Google Scholar] [CrossRef]
- Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef] [Green Version]
- Mocak, J.; Bond, A.M.; Mitchell, S.; Scollary, G. A statistical overview of standard (IUPAC and ACS) and new procedures for determining the limits of detection and quantification: Application to voltammetric and stripping techniques (Technical Report). Pure Appl. Chem. 1997, 69, 297–328. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical methods: Fundamentals and applications. In Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons, Inc: New York, NY, USA, 2000; ISBN 0-471-04372-9. [Google Scholar]
- Licht, S.; Cammarata, V.; Wrighton, M.S. Direct measurements of the physical diffusion of redox active species: Microelectrochemical experiments and their simulation. J. Phys. Chem. 1990, 94, 6133–6140. [Google Scholar] [CrossRef]
- Randles, J.E.B. Kinetics of rapid electrode reactions. Discuss. Faraday Soc. 1947, 1, 11. [Google Scholar] [CrossRef]
- Vedantam, A.; Gopinath, S.P.; Robertson, C.S. Acute Management of Traumatic Brain Injury. In Rehabilitation after Traumatic Brain Injury; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–11. [Google Scholar]
- Chen, D.; Tao, Q.; Liao, L.W.; Liu, S.X.; Chen, Y.X.; Ye, S. Determining the Active Surface Area for Various Platinum Electrodes. Electrocatalysis 2011, 2, 207–219. [Google Scholar] [CrossRef]
- Daubinger, P.; Kieninger, J.; Unmüssig, T.; Urban, G.A. Electrochemical characteristics of nanostructured platinum electrodes-A cyclic voltammetry study. Phys. Chem. Chem. Phys. 2014, 16, 8392–8399. [Google Scholar] [CrossRef] [PubMed]
- Inzelt, G.; Berkes, B.B.; Kriston, Á. Electrochemical nanogravimetric studies of adsorption, deposition, and dissolution processes occurring at platinum electrodes in acid media. Pure Appl. Chem. 2010, 83, 269–279. [Google Scholar] [CrossRef]
- Klimes, P.; Duque, J.J.; Brinkmann, B.; Van Gompel, J.; Stead, M.; St. Louis, E.K.; Halamek, J.; Jurak, P.; Worrell, G. The functional organization of human epileptic hippocampus. J. Neurophysiol. 2016, 115, 3140–3145. [Google Scholar] [CrossRef]
- Szostak, K.M.; Grand, L.; Constandinou, T.G. Neural Interfaces for Intracortical Recording: Requirements, Fabrication Methods, and Characteristics. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Shokoueinejad, M.; Park, D.-W.; Jung, Y.; Brodnick, S.; Novello, J.; Dingle, A.; Swanson, K.; Baek, D.-H.; Suminski, A.; Lake, W.; et al. Progress in the Field of Micro-Electrocorticography. Micromachines 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehler, C.; Stieglitz, T.; Asplund, M. Nanostructured platinum grass enables superior impedance reduction for neural microelectrodes. Biomaterials 2015, 67, 346–353. [Google Scholar] [CrossRef]
- Pennings, F.A.; Schuurman, P.R.; van den Munckhof, P.; Bouma, G.J. Brain Tissue Oxygen Pressure Monitoring in Awake Patients during Functional Neurosurgery: The Assessment of Normal Values. J. Neurotrauma 2008, 25, 1173–1177. [Google Scholar] [CrossRef]
- Johnston, A.J.; Steiner, L.A.; Gupta, A.K.; Menon, D.K. Cerebral oxygen vasoreactivity and cerebral tissue oxygen reactivity. Br. J. Anaesth. 2003, 90, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Schurr, A.; Miller, J.J.; Payne, R.S.; Rigor, B.M. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci. 1999, 19, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Al-Mufti, F.; Lander, M.; Smith, B.; Morris, N.A.; Nuoman, R.; Gupta, R.; Lissauer, M.E.; Gupta, G.; Lee, K. Multimodality Monitoring in Neurocritical Care: Decision-Making Utilizing Direct And Indirect Surrogate Markers. J. Intensive Care Med. 2019, 34, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lin, S.-C.; Nicolelis, M.A.L. Acquiring local field potential information from amperometric neurochemical recordings. J. Neurosci. Methods 2009, 179, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.M.; Laranjinha, J.; Barbosa, R.M.; Sirota, A. Simultaneous measurement of cholinergic tone and neuronal network dynamics in vivo in the rat brain using a novel choline oxidase based electrochemical biosensor. Biosens. Bioelectron. 2015, 69, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço, C.F.; Ledo, A.; Gerhardt, G.A.; Laranjinha, J.; Barbosa, R.M. Neurometabolic and electrophysiological changes during cortical spreading depolarization: Multimodal approach based on a lactate-glucose dual microbiosensor arrays. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Ledo, A.; Lourenço, C.F.; Laranjinha, J.; Gerhardt, G.A.; Barbosa, R.M. Combined in vivo Amperometric Oximetry and Electrophysiology in a Single Sensor—A Tool for Epilepsy Research. Anal. Chem. 2017, 89, acs.analchem.7b03452. [Google Scholar] [CrossRef]
- Viggiano, A.; Marinesco, S.; Pain, F.; Meiller, A.; Gurden, H. Reconstruction of field excitatory post-synaptic potentials in the dentate gyrus from amperometric biosensor signals. J. Neurosci. Methods 2012, 206, 1–6. [Google Scholar] [CrossRef]
- Ledo, A.; Lourenço, C.F.; Laranjinha, J.; Gerhardt, G.A.; Barbosa, R.M. Concurrent measurements of neurochemical and electrophysiological activity with microelectrode arrays: New perspectives for constant potential amperometry. Curr. Opin. Electrochem. 2018, 12, 129–140. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 (Ω) | R2 | Q | n | AW (Ω·s−0.5) | Z at 1 kHz |
|---|---|---|---|---|---|
| 18.98 ± 2.06 | 133.5 ± 33.4 Ω | 47.39 ± 37.99 µF sn−1 | 0.68 ± 0.05 | 257 ± 14 | 77.44 ± 20.55 Ω |
| 9.3 ± 2.2 * Ω cm2 | 0.65 ± 0.49 * mF sn−1 cm−2 | 5.4 ± 1.3 * Ω cm2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledo, A.; Fernandes, E.; Quintero, J.E.; Gerhardt, G.A.; Barbosa, R.M. Electrochemical Evaluation of a Multi-Site Clinical Depth Recording Electrode for Monitoring Cerebral Tissue Oxygen. Micromachines 2020, 11, 632. https://doi.org/10.3390/mi11070632
Ledo A, Fernandes E, Quintero JE, Gerhardt GA, Barbosa RM. Electrochemical Evaluation of a Multi-Site Clinical Depth Recording Electrode for Monitoring Cerebral Tissue Oxygen. Micromachines. 2020; 11(7):632. https://doi.org/10.3390/mi11070632
Chicago/Turabian StyleLedo, Ana, Eliana Fernandes, Jorge E. Quintero, Greg A. Gerhardt, and Rui M. Barbosa. 2020. "Electrochemical Evaluation of a Multi-Site Clinical Depth Recording Electrode for Monitoring Cerebral Tissue Oxygen" Micromachines 11, no. 7: 632. https://doi.org/10.3390/mi11070632