Programmable µChopper Device with On-Chip Droplet Mergers for Continuous Assay Calibration


Abstract
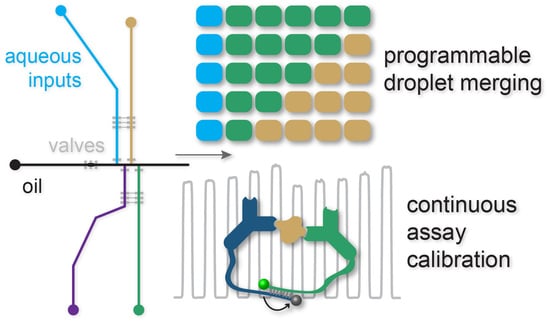
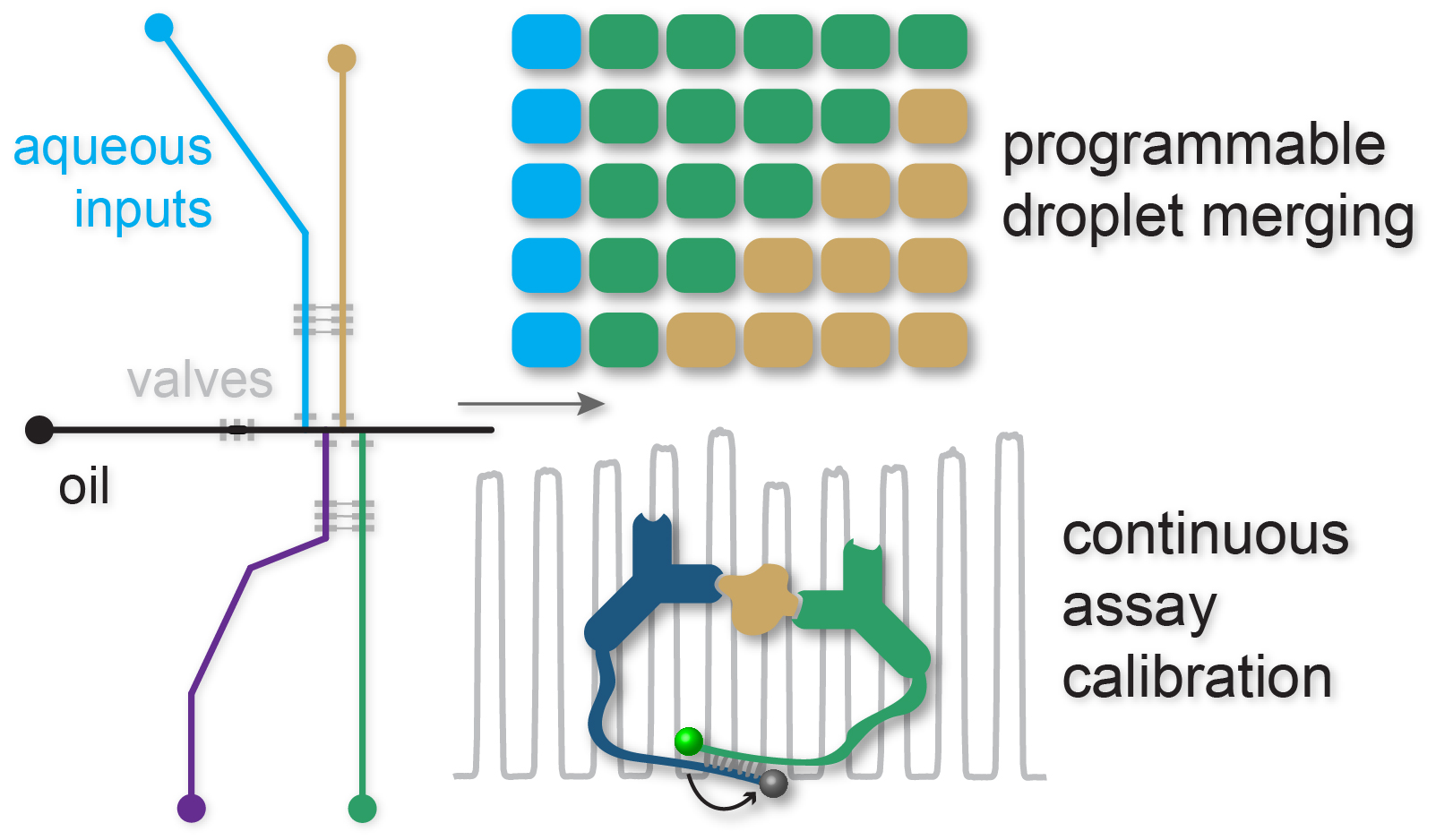
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Equipment
2.2. Microfluidic Master Wafer Fabrication
2.3. Microchip Fabrication
2.4. Flow Control and Droplet Generation
2.5. Programmable Merging of Droplets with Salt Water Electrodes
3. Results and Discussion
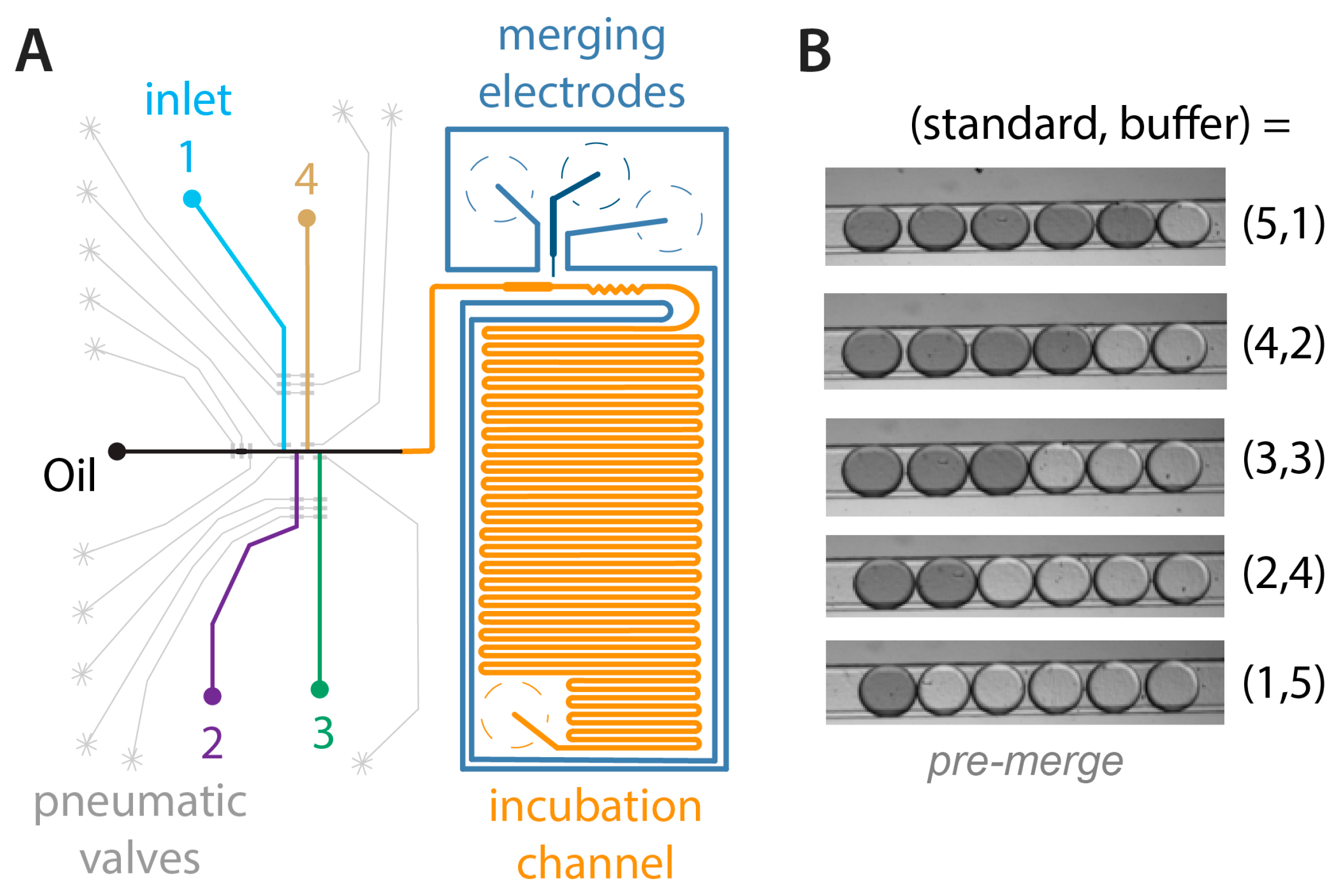
3.1. Microfluidic Device Design and Operation
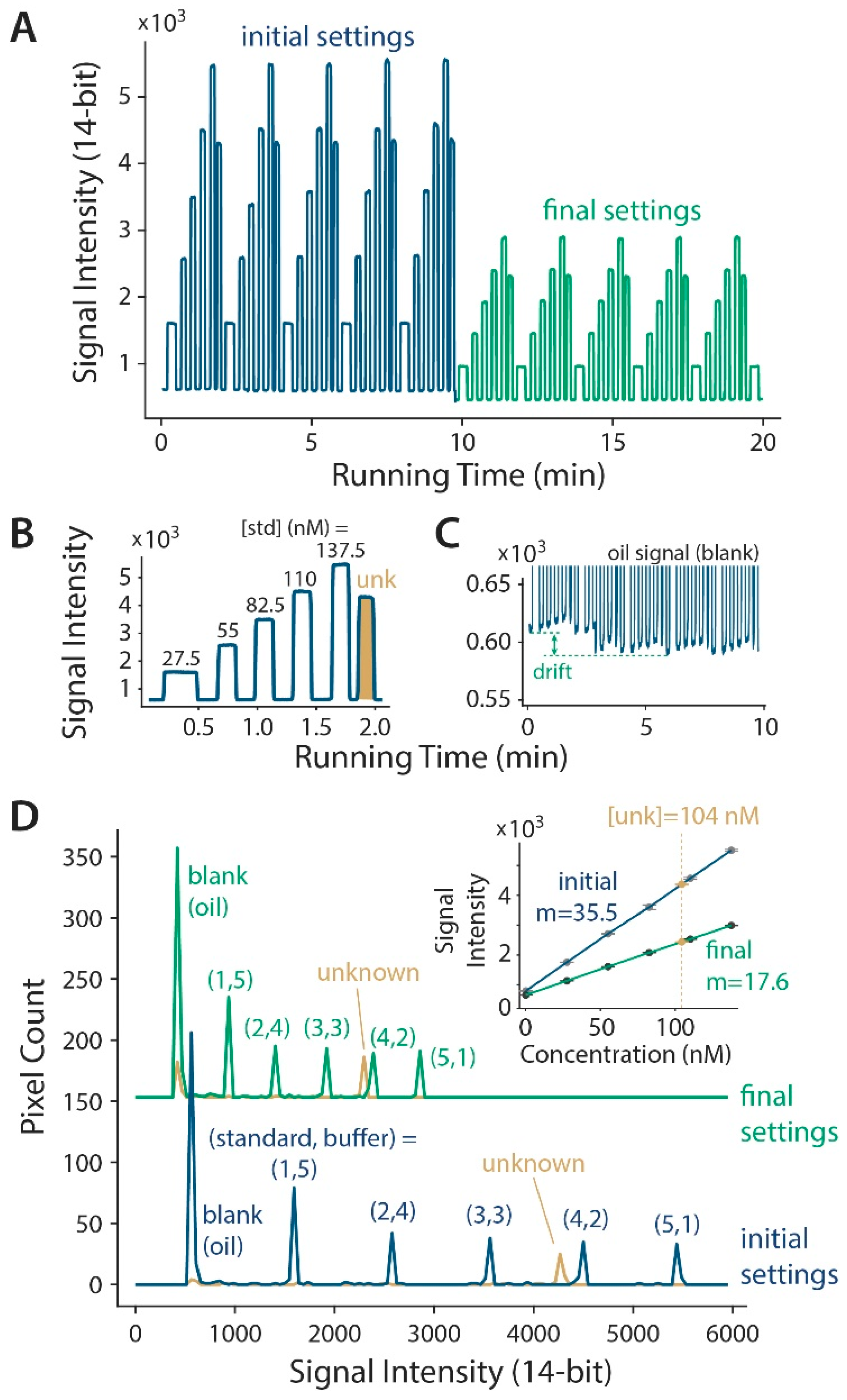
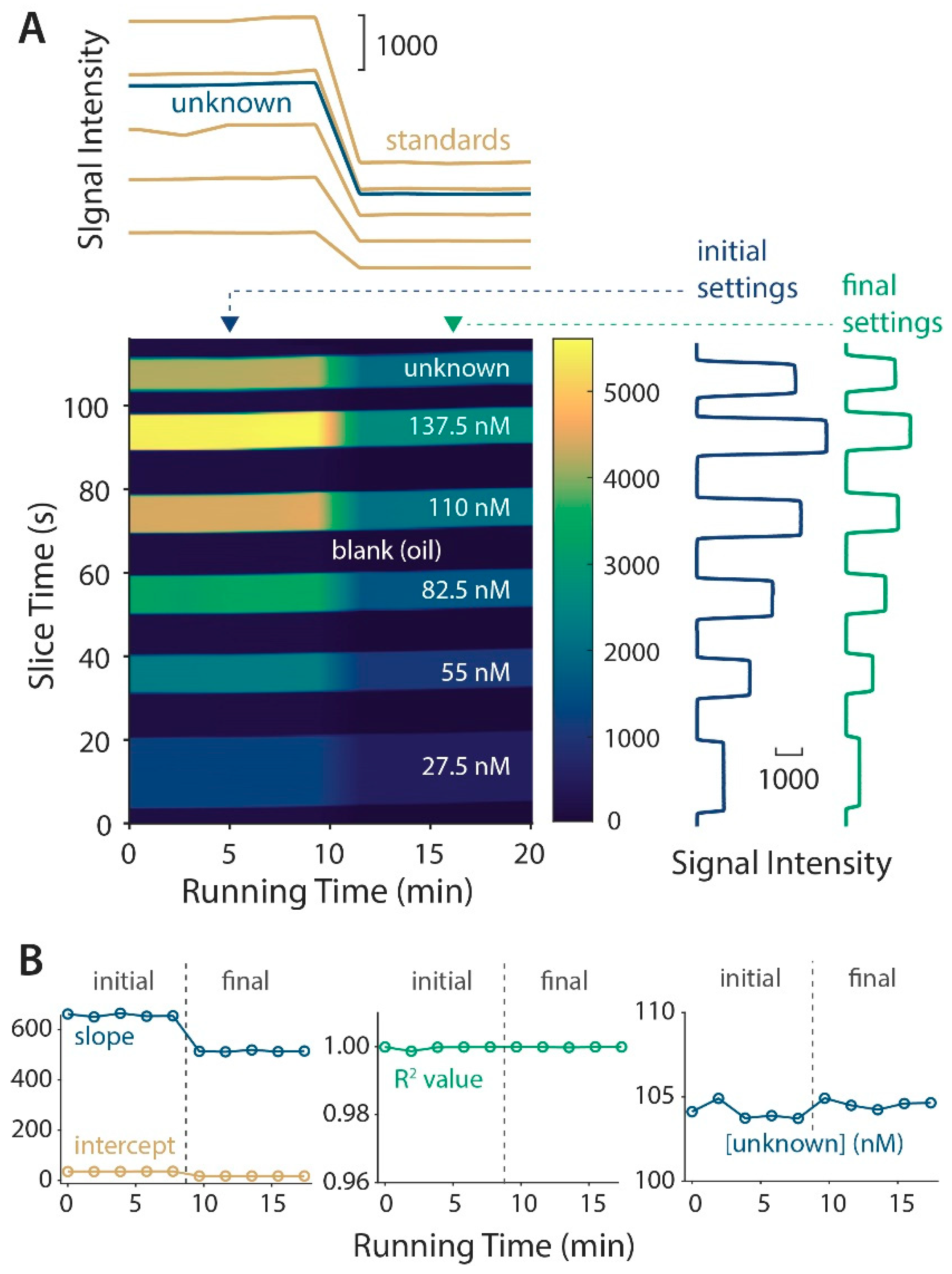
3.2. Microdevice Characterization with Continuous Linear Calibration
3.3. Unique Data Reshaping Using MATLAB Code
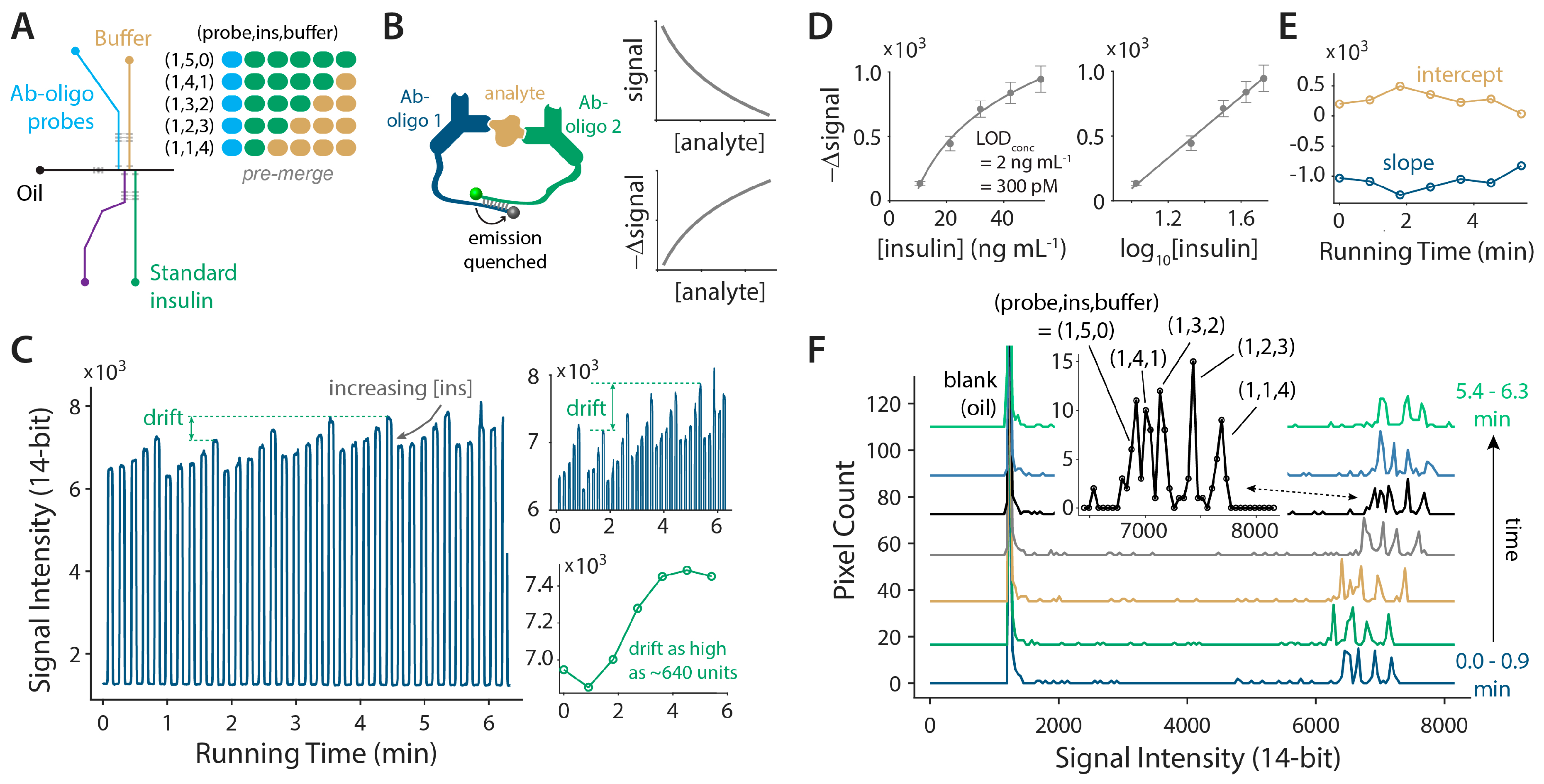
3.4. Continuous Calibration Using a Nonlinear Homogeneous Immunoassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lorenz, R.M.; Fiorini, G.S.; Jeffries, G.D.; Lim, D.S.; He, M.; Chiu, D.T. Simultaneous generation of multiple aqueous droplets in a microfluidic device. Anal. Chim. Acta 2008, 630, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Haliburton, J.R.; Yuan, A.; Abate, A.R. Droplet barcoding for massively parallel single-molecule deep sequencing. Nat. Commun. 2016, 7, 11784. [Google Scholar] [CrossRef] [PubMed]
- Coelho, B.; Veigas, B.; Fortunato, E.; Martins, R.; Águas, H.; Igreja, R.; Baptista, P. Digital microfluidics for nucleic acid amplification. Sensors 2017, 17, 1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Carugo, D.; Crake, C.; Owen, J.; de Saint Victor, M.; Seth, A.; Coussios, C.; Stride, E. Nanoparticle-loaded protein–polymer nanodroplets for improved stability and conversion efficiency in ultrasound imaging and drug delivery. Adv. Mater. 2015, 27, 5484–5492. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.H.; Lee, S.S.; Lee, B.; Jeong, S.G.; Peter, M.; Lee, C.S. Programmable static droplet array for the analysis of cell–cell communication in a confined microenvironment. Anal. Chem. 2017, 89, 9722–9729. [Google Scholar] [CrossRef]
- Terekhov, S.S.; Smirnov, I.V.; Stepanova, A.V.; Bobik, T.V.; Mokrushina, Y.A.; Ponomarenko, N.A.; Belogurov, A.A.; Rubtsova, M.P.; Kartseva, O.V.; Gomzikova, M.O. Microfluidic droplet platform for ultrahigh-throughput single-cell screening of biodiversity. Proc. Natl. Acad. Sci. USA 2017, 114, 2550–2555. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hu, J.; Easley, C.J. Automated microfluidic droplet sampling with integrated, mix-and-read immunoassays to resolve endocrine tissue secretion dynamics. Lab Chip 2018, 18, 2926–2935. [Google Scholar] [CrossRef]
- Negou, J.T.; Avila, L.A.; Li, X.; Hagos, T.M.; Easley, C.J. Automated microfluidic droplet-based sample chopper for detection of small fluorescence differences using lock-in analysis. Anal. Chem. 2017, 89, 6153–6159. [Google Scholar] [CrossRef]
- Hu, J.; Li, X.; Judd, R.L.; Easley, C.J. Rapid lipolytic oscillations in ex vivo adipose tissue explants revealed through microfluidic droplet sampling at high temporal resolution. Lab Chip 2020, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Easley, C.J.; Rocheleau, J.V.; Head, W.S.; Piston, D.W. Quantitative measurement of zinc secretion from pancreatic islets with high temporal resolution using droplet-based microfluidics. Anal. Chem. 2009, 81, 9086–9095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJournette, C.J.; Kim, J.; Medlen, H.; Li, X.; Vincent, L.J.; Easley, C.J. Creating biocompatible oil–water interfaces without synthesis: Direct interactions between primary amines and carboxylated perfluorocarbon surfactants. Anal. Chem. 2013, 85, 10556–10564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deal, K.S.; Easley, C.J. Self-regulated, droplet-based sample chopper for microfluidic absorbance detection. Anal. Chem. 2012, 84, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Sciambi, A.; Abate, A.R. Accurate microfluidic sorting of droplets at 30 khz. Lab Chip 2015, 15, 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Wang, L. Passive and active droplet generation with microfluidics: A review. Lab Chip 2017, 17, 34–75. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Shin, M.; Wang, T. Programmable active droplet generation enabled by integrated pneumatic micropumps. Lab Chip 2013, 13, 267–273. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Tan, S.H.; Gañán-Calvo, A.M.; Tor, S.B.; Loh, N.H.; Nguyen, N.-T. Active droplet generation in microfluidics. Lab Chip 2016, 16, 35–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doonan, S.R.; Lin, M.; Bailey, R.C. Droplet car-wash: Continuous picoliter-scale immunocapture and washing. Lab Chip 2019, 19, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, B.; Eastburn, D.J.; Abate, A.R. Electrode-free picoinjection of microfluidic drops. Lab Chip 2012, 12, 4029–4032. [Google Scholar] [CrossRef]
- Li, X.P.; Brooks, J.C.; Hu, J.; Ford, K.I.; Easley, C.J. 3d-templated, fully automated microfluidic input/output multiplexer for endocrine tissue culture and secretion sampling. Lab Chip 2017, 17, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-T.; Lee, G.-B. Formation of microdroplets in liquids utilizing active pneumatic choppers on a microfluidic chip. J. Microelectromech. Syst. 2006, 15, 1492–1498. [Google Scholar] [CrossRef]
- Godwin, L.A.; Deal, K.S.; Hoepfner, L.D.; Jackson, L.A.; Easley, C.J. Measurement of microchannel fluidic resistance with a standard voltage meter. Anal. Chim. Acta 2013, 758, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negou, J.T.; Hu, J.; Li, X.; Easley, C.J. Advancement of analytical modes in a multichannel, microfluidic droplet-based sample chopper employing phase-locked detection. Anal. Methods 2018, 10, 3436–3443. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Tice, J.D.; Ismagilov, R.F. A microfluidic system for controlling reaction networks in time. Angew. Chem. Int. Ed. 2003, 42, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-K.; Ali, M.M.; Zhang, K.; Huang, S.S.; Peterson, E.; Digman, M.A.; Gratton, E.; Zhao, W. Rapid detection of single bacteria in unprocessed blood using integrated comprehensive droplet digital detection. Nat. Commun. 2014, 5, 5427. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.M.; Sousa, K.M.; Jennings, C.; MacDougald, O.A.; Kennedy, R.T. Continuous-flow enzyme assay on a microfluidic chip for monitoring glycerol secretion from cultured adipocytes. Anal. Chem. 2009, 81, 2350–2356. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Gulati, S.; Edel, J.B.; Demello, A.J. Pillar-induced droplet merging in microfluidic circuits. Lab Chip 2008, 8, 1837–1841. [Google Scholar] [CrossRef]
- Sander, J.S.; Erb, R.M.; Denier, C.; Studart, A.R. Magnetic transport, mixing and release of cargo with tailored nanoliter droplets. Adv. Mater. 2012, 24, 2582–2587. [Google Scholar] [CrossRef]
- Sciambi, A.; Abate, A.R. Generating electric fields in pdms microfluidic devices with salt water electrodes. Lab Chip 2014, 14, 2605–2609. [Google Scholar] [CrossRef]
- Sesen, M.; Alan, T.; Neild, A. Microfluidic on-demand droplet merging using surface acoustic waves. Lab Chip 2014, 14, 3325–3333. [Google Scholar] [CrossRef]
- Doonan, S.R.; Bailey, R.C. K-channel: A multifunctional architecture for dynamically reconfigurable sample processing in droplet microfluidics. Anal. Chem. 2017, 89, 4091–4099. [Google Scholar] [CrossRef] [Green Version]
- Erickstad, M.; Gutierrez, E.; Groisman, A. A low-cost low-maintenance ultraviolet lithography light source based on light-emitting diodes. Lab Chip 2015, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Heyduk, E.; Dummit, B.; Chang, Y.-H.; Heyduk, T. Molecular pincers: Antibody-based homogeneous protein sensors. Anal. Chem. 2008, 80, 5152–5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Easley, C.J. Homogeneous assays of second messenger signaling and hormone secretion using thermofluorimetric methods that minimize calibration burden. Anal. Chem. 2017, 89, 8517–8523. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, N.; Easley, C.J. Programmable µChopper Device with On-Chip Droplet Mergers for Continuous Assay Calibration. Micromachines 2020, 11, 620. https://doi.org/10.3390/mi11060620
Shi N, Easley CJ. Programmable µChopper Device with On-Chip Droplet Mergers for Continuous Assay Calibration. Micromachines. 2020; 11(6):620. https://doi.org/10.3390/mi11060620
Chicago/Turabian StyleShi, Nan, and Christopher J. Easley. 2020. "Programmable µChopper Device with On-Chip Droplet Mergers for Continuous Assay Calibration" Micromachines 11, no. 6: 620. https://doi.org/10.3390/mi11060620