
Production of CO2 Hydrates in Aqueous Mixtures Having (NH4)2SO4 at Different Concentrations; Definition of Consequences on the Process Evolution, Quantification of CO2 Captured and Validation of Hydrates Production as Technique for Ammonium Removal from Waste Water
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Apparatus
2.2. Materials
2.3. Experimental Procedure
- -
- 1.9 wt%, corresponding to 0.3 wt% (NH4)2SO4 in the aqueous solution;
- -
- 6.3 wt%, corresponding to 1.0 wt% (NH4)2SO4 in the aqueous solution;
- -
- 9.5 wt%, corresponding to 1.5 wt% (NH4)2SO4 in the aqueous solution.
3. Results and Discussion
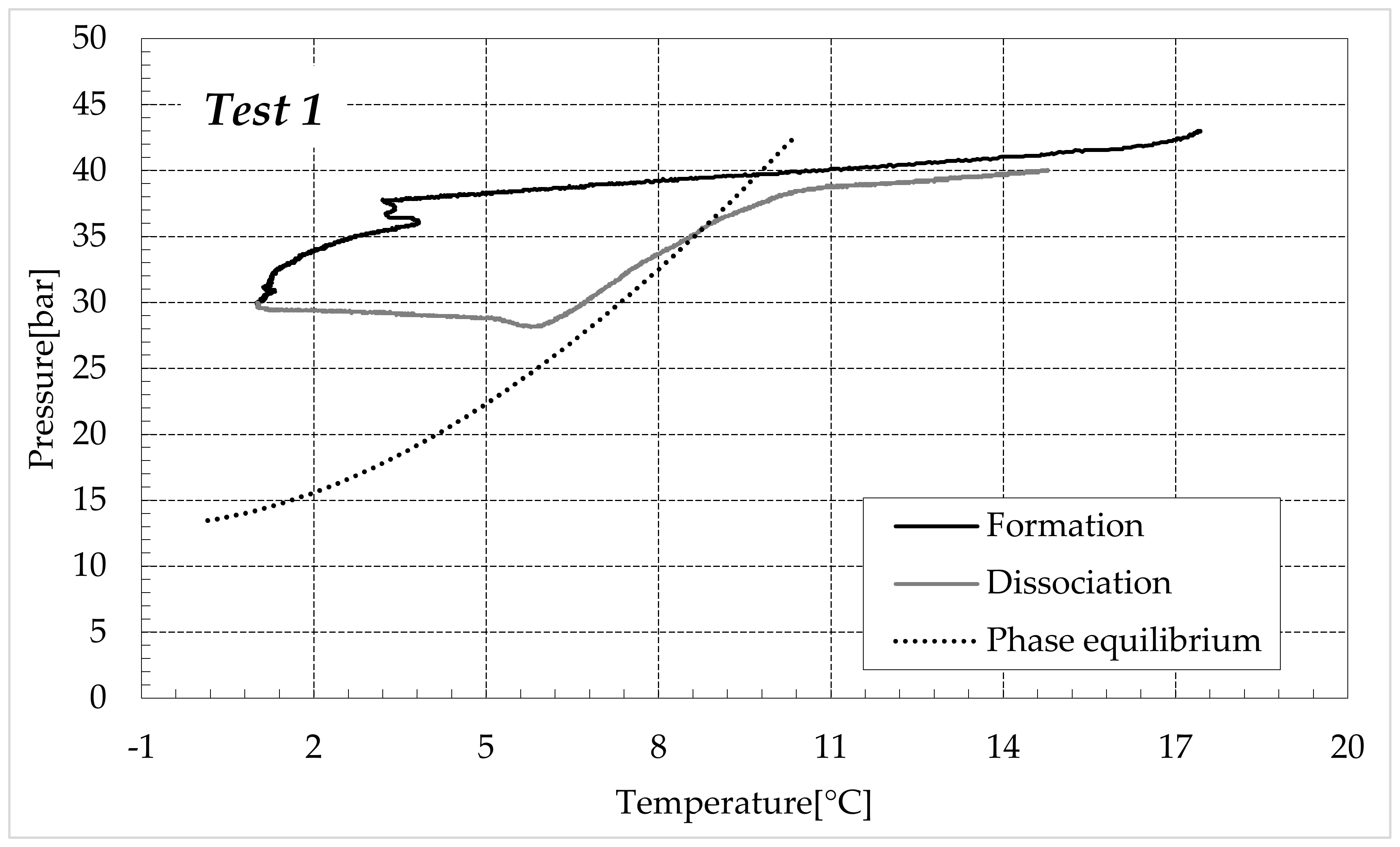
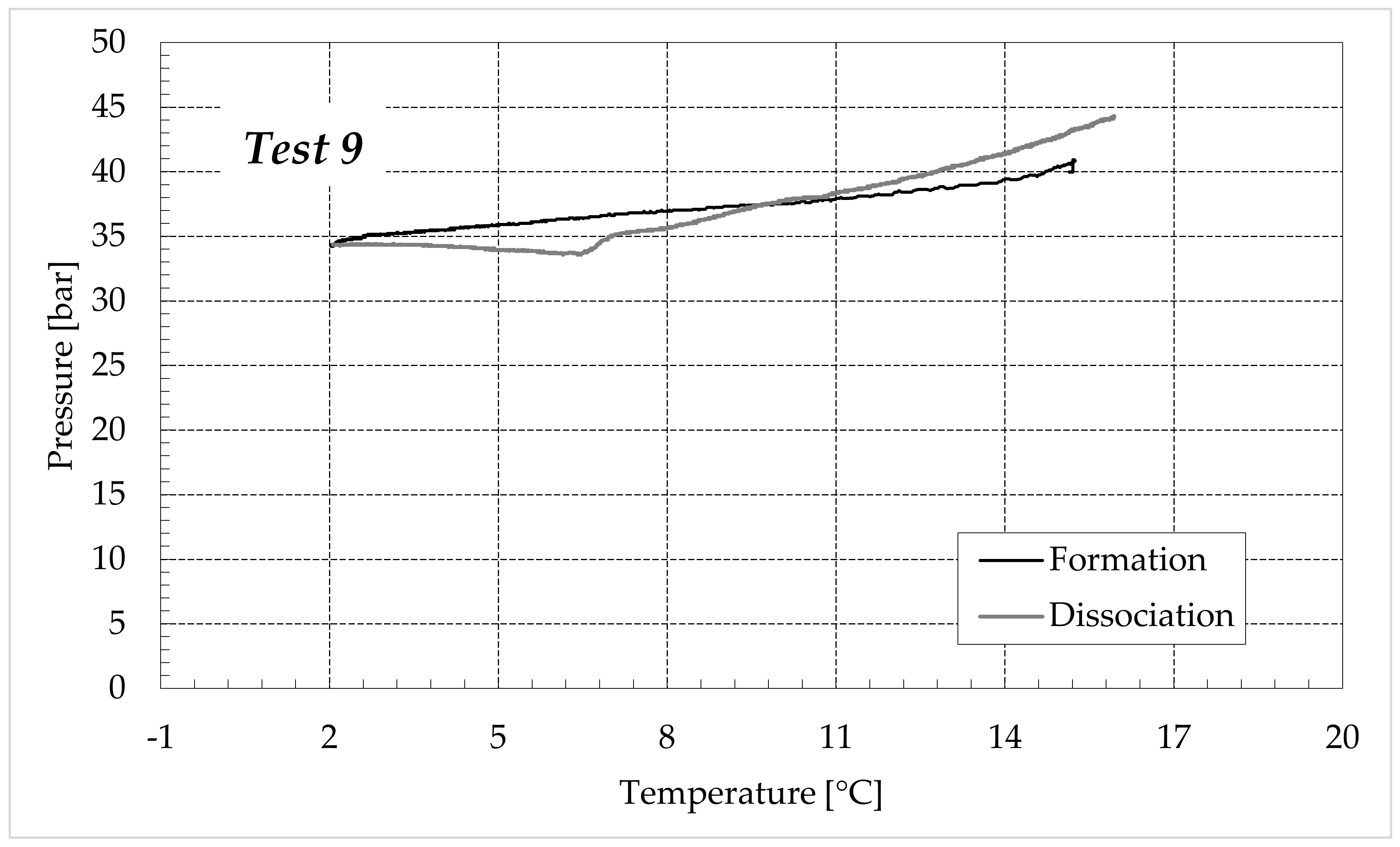
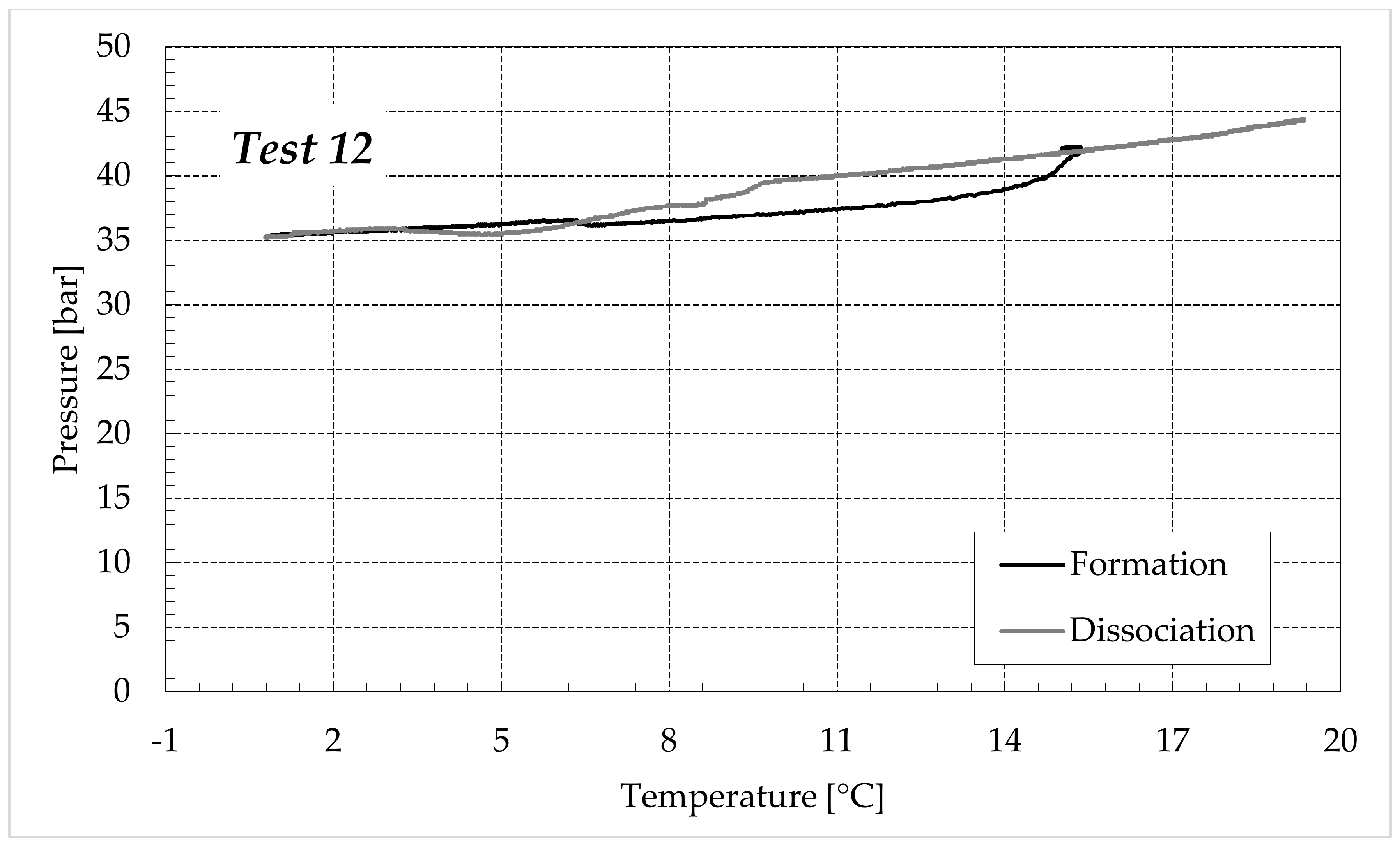
3.1. Formation and Dissociation of CO2 Hydrates in Fresh Water and in Mixtures Containing (NH4)SO4
- -
- Reduction in the presence of 1.9 wt% (NH4)2SO4: from 14.29 to 53.25%;
- -
- Reduction in the presence of 6.3 wt% (NH4)2SO4: from 20.34 to 56.28%;
- -
- Reduction in the presence of 9.5 wt% (NH4)2SO4: from 23.38 to 60.17%.
3.2. Determination of (NH4)SO4 Content in Hydrate Phase, via Spectrophotometric Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galloway, T.J.; Ruska, W.; Chappelear, P.S.; Kobayashi, R. Experimental measurement of hydrate numbers for methane and ethane and comparison with theoretical values. Ind. Eng. Chem. Fundam. 1970, 9, 237–243. [Google Scholar] [CrossRef]
- Englezos, P. Clathrate hydrates. Ind. Eng. Chem. Res. 1993, 32, 1251–1274. [Google Scholar] [CrossRef]
- Sloan, E.D.; Koh, C.A. Clathrate Hydrates on Natural Gases, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Makogon, Y.F.; Holditch, S.A.; Makogon, T.Y. Natural gas hydrates—A potential energy source for the 21st Century. J. Pet. Sci. Eng. 2007, 56, 14–31. [Google Scholar] [CrossRef]
- Makogon, Y.F. Natural gas hydrates—A promising source of energy. J. Nat. Gas Sci. Eng. 2010, 2, 49–59. [Google Scholar] [CrossRef]
- Li, Y.; Gambelli, A.M.; Rossi, F.; Mei, S. Effect of promoters on CO2 hydrate formation: Thermodynamic assessment and microscale Raman spectroscopy/hydrate crystal morphology characterization analysis. Fluid Phase Equilibr. 2021, 550, 113218. [Google Scholar] [CrossRef]
- Ripmeester, J.A.; Tse, J.S.; Ratcliffe, C.I.; Powell, B.M. A new clathrate hydrate structure. Nature 1987, 325, 135–136. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, D.; Yin, J.; Zhou, X.; Li, Y.; Chi, P.; Han, Y.; Ansari, U.; Cheng, Y. Sediment instability caused by gas production from hydrate-bearing sediment in Northern South China Sea by horizontal wellbore: Evolution and mechanism. Nat. Resour. Res. 2023. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J. Factors affecting the lower limit of the safe mud weight window for drilling operation in hydrate-bearing sediments in the Northern South China Sea. Geophys. Geo-Energy Geo-Resour. 2022, 8, 82. [Google Scholar] [CrossRef]
- Wei, W.N.; Li, B.; Gan, Q.; Li, Y.L. Research progress of natural gas hydrate exploitation with CO2 replacement: A review. Fuel 2022, 312, 122873. [Google Scholar] [CrossRef]
- Kvenvolden, K.A. Gas hydrates—Geological perspective and global change. Rev. Geophys. 1993, 31, 173–187. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Re-definition of the region suitable for CO2/CH4 replacement into hydrates as a function of the thermodynamic difference between CO2 hydrate formation and dissociation. Process Saf. Environ. Prot. 2023, 169, 132–141. [Google Scholar] [CrossRef]
- Rossi, F.; Gambelli, A.M.; Sharma, D.K.; Castellani, B.; Nicolini, A.; Castaldi, M.J. Experiments on methane hydrates formation in seabed deposits and gas recovery adopting carbon dioxide replacement strategies. Appl. Therm. Eng. 2019, 148, 371–381. [Google Scholar] [CrossRef]
- Li, X.Y.; Li, X.S.; Wang, Y.; Liu, J.W.; Hu, H.Q. The optimization mechanism for gas hydrate dissociation by depressurization in the sediment with different water saturations and different particle sizes. Energy 2021, 215, 119129. [Google Scholar] [CrossRef]
- Xu, C.G.; Cai, J.; Yu, Y.S.; Chen, Z.Y.; Li, X.S. Research on micro-mechanism and efficiency of CH4 exploitation via CH4-CO2 replacement from natural gas hydrates. Fuel 2018, 216, 255–265. [Google Scholar] [CrossRef]
- Tung, Y.T.; Chen, L.J.; Chen, Y.P.; Lin, S.T. In situ methane recovery and carbon dioxide sequestration in methane hydrates: A molecular dynamics simulation study. J. Phys. Chem. B 2011, 115, 15295–15302. [Google Scholar] [CrossRef]
- Baldwin, B.A.; Stevens, J.; Howard, J.J.; Graue, A.; Kvamme, B.; Aspenes, E.; Ersland, G.; Husebo, J.; Zornes, D.R. Using magnetic resonance imaging to monitor CH4 hydrate formation and spontaneous conversion of CH4 hydrate to CO2 hydrate in porous media. Magn. Reson. Imaging 2009, 27, 720–726. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Stornelli, G.; Di Schino, A.; Rossi, F. Methane and carbon dioxide hydrates properties in presence of Inconel 718 particles: Analyses on its potential application in gas separation processes to perform efficiency improvement. J. Env. Chem. Eng. 2021, 9, 106571. [Google Scholar] [CrossRef]
- Ren, J.; Zeng, S.; Chen, D.; Yang, M.; Linga, P.; Yin, Z. Roles of montmorillonite clay on the kinetics and morphology of CO2 hydrate in hydrate-based CO2 sequestration. Appl. Energy 2023, 340, 120997. [Google Scholar] [CrossRef]
- Liu, Z.; Zheng, J.; Wang, Z.; Gao, Y.; Sun, B.; Liao, Y.; Linga, P. Effect of clay on methane hydrate formation and dissociation in sediment: Implications for energy recovery from clayey-sandy hydrate reservoirs. Appl. Energy 2023, 341, 121064. [Google Scholar] [CrossRef]
- Koide, H.; Shindo, Y.; Tazaki, Y.; Iijima, M.; Ito, K.; Kimura, N.; Omata, K. Deep sub-seabed disposal of CO2—The most protective storage. Energy Convers. Manag. 1997, 38, 253–258. [Google Scholar] [CrossRef]
- Ganteda, R.R.; Burla, S.K.; Boggu, J.M.R.; Prasad, P.S.R. Efficient storage of methane in hydrate from using soybean powder. Methane 2022, 1, 201–209. [Google Scholar] [CrossRef]
- Kumar, S.; Kwon, H.T.; Choi, K.H.; Lim, W.; Cho, J.H.; Tak, K.; Moon, L. LNG: An eco-friendly cryogenic fuel for sustainable development. Appl. Energy 2011, 88, 4264–4273. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, Y.J.; Zheng, T.; Yuan, Q.; Sun, C.Y.; Yang, L.Y.; Chen, G.J. Replacement in CH4-CO2 hydrate below freezing point based on abnormal self-preservation differences of CH4 hydrate. Chem. Eng. J. 2021, 403, 126283. [Google Scholar] [CrossRef]
- Sun, Z.G.; Wang, R.; Ma, R.; Guo, K.; Fan, S. Natural gas storage in hydrates with the presence of promoters. Energy Convers. Manag. 2003, 44, 2733–2742. [Google Scholar] [CrossRef]
- House, K.Z.; Schrag, D.P.; Harvey, C.F.; Lackner, K.S. Permanent carbon dioxide storage in deep-sea sediments. Proc. Natl. Acad. Sci. USA 2006, 103, 12291–12295. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, S.; Cheng, Z.; Huang, M.; Zhang, Y.; Zheng, J.; Jiang, L.; Liu, Y. Dependence of the hydrate-based CO2 storage processes on the hydrate reservoir environment in high-efficiency storage methods. Chem. Eng. J. 2021, 415, 128937. [Google Scholar] [CrossRef]
- Wang, F.; Guo, G.; Liu, G.Q.; Luo, S.J.; Guo, R.B. Effects of surfactants micelles and surfactant-coated nanospheres on methane hydrate growth pattern. Chem. Eng. Sci. 2016, 144, 108–115. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Rossi, F. Review on the usage of small-chain hydrocarbons (C2-C4) as adi gases for improving the efficiency of hydrate-based technologies. Energies 2023, 16, 3576. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Wong, A.J.H.; Babu, P.; Kumar, R.; Kulprathipanja, S.; Rangsungivit, P.; Linga, P. Rapid methane hydrate formation to develop a cost effective large scale energy storage system. Chem. Eng. J. 2016, 290, 161–173. [Google Scholar] [CrossRef]
- Montazeri, S.M.; Kolliopoulos, G. Hydrate based desalination for sustainable water treatment: A review. Desalination 2022, 537, 115855. [Google Scholar] [CrossRef]
- Bhatti, Z.A.; Qureshi, K.; Maitlo, G.; Ahmed, S. Study of PAN fiber and iron ore absorbents for arsenic removal. Civ. Eng. J. 2020, 6, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Priscilla, S.J.; Judi, V.A.; Daniel, R.; Sivaji, K. Effects of chromium doping on the electrical properties of ZnO nanoparticles. Emerg. Sci. J. 2020, 4, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, J.; Gambelli, A.M.; Zhao, X.; Gao, Y.; Rossi, F.; Mei, S. In situ experimental study on the effect of mixed inhibitors on the phase equilibrium of carbon dioxide hydrate. Chem. Eng. Sci. 2022, 248, 117230. [Google Scholar] [CrossRef]
- Bates, C.; Gladwin, R.; McGrath, L. Desalination in pilot scale column crystallizers. Desalination 1977, 21, 83–97. [Google Scholar] [CrossRef]
- Karamoddin, M.; Varaminian, F. Water desalination using R141b gas hydrate formation. Desalination Water Treat. 2014, 52, 2450–2456. [Google Scholar] [CrossRef]
- Chong, Z.R.; Yang, S.H.B.; Babu, P.; Linga, P.; Li, X.S. Review on natural gas hydrate as an energy resource: Prospects and challenges. Appl. Energy 2016, 162, 1633–1652. [Google Scholar] [CrossRef]
- Bradshaw, R.W.; Simmons, B.A.; Majzoub, E.H.; Clift, W.M.; Dedrick, D.E. Clathrate hydrates for production of potable water. Mater. Res. Soc. Symp. Proc. 2006, 930, 14–19. [Google Scholar] [CrossRef]
- Bavoh, C.B.; Partoon, B.; Lal, B.; Gonfa, G.; Foo Khor, S.; Sharif, A.M. Inhibition effect of amino acids on carbon dioxide hydrate. Chem. Eng. Sci. 2017, 171, 331–339. [Google Scholar] [CrossRef]
- Gambelli, A.M.; Filipponi, M.; Nicolini, A.; Rossi, F. Natural gas hydrate: Effect of sodium chloride on the CO2 replacement process. Int. Multidiscip. Sci. GeoConference SGEM 2019, 19, 333–343. [Google Scholar]
- Holzammer, C.; Finckenstein, A.; Will, S.; Braeuer, A.S. How sodium chloride salt inhibits the formation of CO2 gas hydrates. J. Phys. Chem. B 2016, 120, 2452–2459. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.H.; Seol, Y. Increasing gas hydrate formation temperature for desalination of high salinity produced water with secondary guests. ACS Sustain. Chem. Eng. 2013, 1, 1218–1224. [Google Scholar] [CrossRef]
- Sahu, P.; Krishnaswamy, K.; Ponnani, N.K.; Pande, A. A thermodynamic approach to selection of suitable hydrate formers for seawater desalination. Desalination 2018, 436, 144–151. [Google Scholar] [CrossRef]
- Chapoy, A.; Mazloum, S.; Burgass, R.; Haghighi, H.; Tohidi, B. Clathrate hydrate equilibria in mixed monoethylene glycol and electrolyte aqueous solutions. J. Chem. Thermodyn. 2012, 48, 7–12. [Google Scholar] [CrossRef]
- Gaikwad, N.; Nakka, R.; Khavala, V.; Bhadani, A.; Mamane, H.; Kumar, R. Gas hydrate-based process for desalination of heavy metal ions from an aqueous solution: Kinetics and rate of recovery. ACS EST Water 2021, 1, 134–144. [Google Scholar] [CrossRef]
- Nam Park, K.; Hong, S.Y.; Lee, J.W.; Kang, K.C.; Lee, Y.C.; Ha, M.G.; Lee, J.D. A new apparatus for seawater desalination by gas hydrate process and removal characteristics of dissolved minerals (Na+, Mg2+, Ca2+, K+, B3+). Desalination 2011, 274, 91–96. [Google Scholar] [CrossRef]
- Lu, H.; Matsumoto, R.; Tsuji, Y.; Oda, H. Anion plays a more important role than cation in affecting gas hydrate stability in electrolyte solutions?—A recognition from experimental results. Fluid Phase Equilibr. 2001, 178, 225–232. [Google Scholar] [CrossRef]
- Englezos, P.; Kalogerakis, N.; Dholabhai, P.D.; Bishnoi, P.R. Kinetics of formation of methane and ethane gas hydrates. Chem. Eng. Sci. 1987, 42, 2647–2658. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, X.; Shaheen, S.M.; Zhao, Q.; Liu, X.; Rinklebe, J.; Ren, H. Ammonium nitrogen recovery from digestate by hydrothermal pretreatment followed by activated hydrochar sorption. Chem. Eng. J. 2020, 379, 122254. [Google Scholar] [CrossRef]
- Zhang, N.; Zheng, H.Y.; Hu, X.H.; Zhu, Q.; Stanislaus, M.S.; Li, S.Y.; Zhao, C.Y.; Wang, Q.H.; Yang, Y.N. Enhanced bio—Methane production from ammonium—Rich waste using eggshell- and lignite- modified zeolite (ELMZ) as a bio—Adsorbent during anaerobic digestion. Process Biochem. 2019, 81, 148–155. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Q.C.; Ding, L.L.; Ren, H.Q.; Xu, K.; Wu, Y.G.; Sheng, D. Modeling assessment for ammonium nitrogen recovery from wastewater by chemical precipitation. J. Environ. Sci. 2011, 23, 881–890. [Google Scholar] [CrossRef]
- Mandal, S.; Thangarajan, R.; Bolan, N.S.; Sarkar, B.; Khan, N.; Ok, Y.S.; Naidu, R. Biochar—Induced concomitant decrease in ammonia volatilization and increase in nitrogen use efficiency be wheat. Chemosphere 2016, 142, 120–127. [Google Scholar] [CrossRef]
- Takaya, C.A.; Fletcher, L.A.; Singh, S.; Anyikude, K.U.; Boss, A.B. Phosphate and ammonium sorption capacity of biochar and hydrochar from different wastes. Chemosphere 2016, 145, 518–527. [Google Scholar] [CrossRef]
- Ajmal, S.; Kumar, A.; Selvaraj, M.; Alam, M.M.; Yang, Y.; Das, D.K.; Gupta, R.K.; Yasin, G. MXenes and their interfaces for the taming of carbon dioxide & nitrate: A critical review. Coord. Chem. Rev. 2023, 483, 215094. [Google Scholar]
- Ajmal, S.; Yasin, G.; Kumar, A.; Tabish, M.; Ibraheem, S.; Sammed, K.A.; Mushtaq, M.S.; Saad, A.; Mo, Z.; Zhao, W. A disquisition on CO2 electroreduction to C2H4: An engineering and design perspective looking beyond novel choosy catalyst materials. Coord. Chem. Rev. 2023, 485, 215099. [Google Scholar] [CrossRef]
- Rossi, F.; Li, Y.; Gambelli, A.M. Thermodynamic and kinetic description of the main effects related to the memory effect during carbon dioxide hydrates formation in a confined environment. Sustainability 2021, 13, 13797. [Google Scholar] [CrossRef]
- Khan, M.S.; Partoon, B.; Bavoh, C.B.; Lal, B.; Mellon, B.M. Influence of tetramethylammonium hydroxide on methane and carbon dioxide phase equilibrium conditions. Fluid Phase Equilibr. 2017, 440, 1–8. [Google Scholar] [CrossRef]
- Sadeq, D.; Iglauer, S.; Lebedev, M.; Smith, C.; Barifcani, A. Experimental determination of hydrate phase equilibrium for different gas mixture containing methane, carbon dioxide and nitrogen with motor current measurements. J. Nat. Gas Sci. Eng. 2017, 38, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.H.; Zhou, S.D.; Li, X.S.; Wang, S.L. Effect of graphite nanoparticles on CO2 hydrate phase equilibrium. Fluid Phase Equilibr. 2016, 414, 23–28. [Google Scholar] [CrossRef]
- Jarrahian, A.; Nakhaee, A. Hydrate-liquid-vapor equilibrium condition for N2 + CO2 + H2O system: Measurement and modelling. Fuel 2019, 237, 769–774. [Google Scholar] [CrossRef]
- Kyung, D.; Lee, K.; Kim, H.; Lee, W. Effect of marine environmental factors on the phase equilibrium of CO2 hydrate. Int. J. Greenh. Gas Control 2014, 20, 285–292. [Google Scholar] [CrossRef]
- Seo, Y.T.; Lee, H. Multiple-phase hydrate equilibria of the ternary carbon dioxide, methane, and water mixtures. J. Phys. Chem. B 2001, 105, 10084–10090. [Google Scholar] [CrossRef]
- Fitzgerald, G.C.; Castaldi, M.J.; Zhou, Y. Large scale reactor details and results for the formation and decomposition of methane hydrates via thermal stimulation dissociation. J. Pet. Sci. Eng. 2012, 94, 19–27. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (NH4)SO4 in Water [wt%] | Corr. NH4+ in Water [wt%] | Test n° |
|---|---|---|
| 0 | 0 | 1 |
| 2 | ||
| 3 | ||
| 4 | ||
| 1.9 | 0.3 | 5 |
| 6 | ||
| 7 | ||
| 8 | ||
| 6.3 | 1.0 | 9 |
| 10 | ||
| 11 | ||
| 12 | ||
| 9.5 | 1.5 | 13 |
| 14 | ||
| 15 | ||
| 16 |
| Final Pressure Values [bar] | |||
|---|---|---|---|
| Tests 1–4 | Tests 5–8 | Tests 9–12 | Tests 13–16 |
| 0 wt% NH4+ | 0.3 wt% NH4+ | 1.0 wt% NH4+ | 1.5 wt% NH4+ |
| 28.18 | 32.91 | 33.62 | 35.77 |
| 29.83 | 33.17 | 35.63 | 35.64 |
| 27.9 | 32.99 | 33.64 | 35.64 |
| 28.09 | 32.85 | 35.22 | 35.78 |
| Moles of Hydrates Formed [mol] | |||
|---|---|---|---|
| Tests 1–4 | Tests 5–8 | Tests 9–12 | Tests 13–16 |
| 0 wt% NH4+ | 0.3 wt% NH4+ | 1.0 wt% NH4+ | 1.5 wt% NH4+ |
| 0.237 | 0.198 | 0.101 | 0.175 |
| 0.204 | 0.108 | 0.183 | 0.092 |
| 0.256 | 0.140 | 0.184 | 0.177 |
| 0.228 | 0.108 | 0.110 | 0.132 |
| Test | Water into Hydrates [mol] | NH4+ in Water [wt%] |
|---|---|---|
| A | 0.624 | 0.428 |
| B | 0.786 | 0.407 |
| C | 0.588 | 0.400 |
| D | 0.702 | 0.449 |
| E | 0.834 | 0.380 |
| F | 0.732 | 0.428 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambelli, A.M.; Rushani, X.; Pezzolla, D.; Rossi, F.; Gigliotti, G. Production of CO2 Hydrates in Aqueous Mixtures Having (NH4)2SO4 at Different Concentrations; Definition of Consequences on the Process Evolution, Quantification of CO2 Captured and Validation of Hydrates Production as Technique for Ammonium Removal from Waste Water. Sustainability 2023, 15, 9841. https://doi.org/10.3390/su15129841
Gambelli AM, Rushani X, Pezzolla D, Rossi F, Gigliotti G. Production of CO2 Hydrates in Aqueous Mixtures Having (NH4)2SO4 at Different Concentrations; Definition of Consequences on the Process Evolution, Quantification of CO2 Captured and Validation of Hydrates Production as Technique for Ammonium Removal from Waste Water. Sustainability. 2023; 15(12):9841. https://doi.org/10.3390/su15129841
Chicago/Turabian StyleGambelli, Alberto Maria, Xhino Rushani, Daniela Pezzolla, Federico Rossi, and Giovanni Gigliotti. 2023. "Production of CO2 Hydrates in Aqueous Mixtures Having (NH4)2SO4 at Different Concentrations; Definition of Consequences on the Process Evolution, Quantification of CO2 Captured and Validation of Hydrates Production as Technique for Ammonium Removal from Waste Water" Sustainability 15, no. 12: 9841. https://doi.org/10.3390/su15129841