
Development of Green Methods for the Determination of Elemental Impurities in Commercial Pharmaceutical Tablets

 , ,
, ,
Abstract
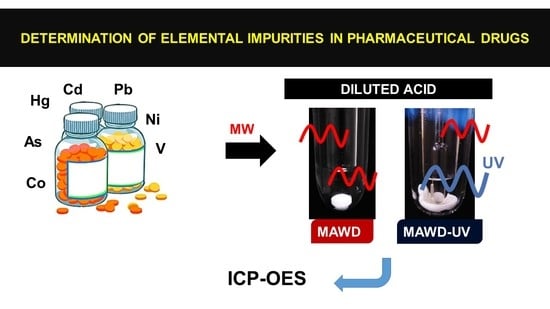
:
1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. Samples, Reagents and Standards
2.3. Sample Preparation Methods
2.3.1. MAWD Method
2.3.2. Evaluation of Digesting Solution
2.3.3. Evaluation of the Irradiation Program
2.3.4. Evaluation of Simultaneous Cooling during Irradiation
2.3.5. Evaluation of the Auxiliary Reagent
2.4. MAWD-UV Method
Evaluation of MAWD-UV Experimental Parameters
2.5. Statistical Treatments
3. Results and Discussion
3.1. Evaluation of Digesting Solution for MAWD
3.2. Evaluation of the Irradiation Program
3.3. Evaluation of Simultaneous Cooling during Irradiation
3.4. Evaluation of Diluted Acid and Auxiliary Reagent for CANA Decomposition
3.5. Concentration of Major Elements in the Samples
3.6. Evaluation of MAWD-UV
3.7. Analytical Figures of Merit of the Proposed Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICH Guideline for Elemental Impurities Q3D(R1). Available online: https://www.ich.org/products/guidelines/quality/quality-single/article/guideline-for-elemental-impurities-copy-1.html (accessed on 7 June 2021).
- Elemental impurities–procedures. In The United States Pharmacopeia, 40th ed.; United States Pharmacopeial: Rockville, MD, USA, 2017.
- Elemental impurities–limits. In The United States Pharmacopeia, 40th ed.; United States Pharmacopeial: Rockville, MD, USA, 2017.
- Peixoto, N.C.; Serafim, M.A.; Flores, E.M.M.; Bebianno, M.J.; Pereira, M.E. Metallothionein, zinc, and mercury levels in tissues of young rats exposed to zinc and subsequently to mercury. Life Sci. 2007, 81, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Barin, J.S.; Mello, P.A.; Mesko, M.F.; Duarte, F.A.; Flores, E.M.M. Determination of elemental impurities in pharmaceutical products and related matrices by ICP-based methods: A review. Anal. Bioanal. Chem. 2016, 408, 4547–4566. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; Fernandes, J.D.R.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Kahn, C.R. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994, 43, 1066–1085. [Google Scholar] [CrossRef]
- Stumvoll, M.; Goldstein, B.J.; Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Menoutis, J.; Parisi, A.; Verma, N. Study of the use of axial viewed inductively coupled plasma atomic emission spectrometry with ultrasonic nebulization for the determination of select elemental impurities in oral drug products. J. Pharm. Biomed. 2018, 152, 12–16. [Google Scholar] [CrossRef]
- Silva, C.S.; Pinheiro, F.C.; Amaral, C.D.B.; Nobrega, J.A. Determination of As, Cd, Hg and Pb in continuous use drugs and excipients by plasma-based techniques in compliance with the United States Pharmacopeia requirements. Spectrochim. Acta Part B At. Spectrosc. 2017, 138, 14–17. [Google Scholar] [CrossRef]
- Støving, C.; Jensen, H.; Gammelgaard, B.; Stürup, S. Development and validation of an ICP-OES method for quantitation of elemental impurities in tablets according to coming US pharmacopeia chapters. J. Pharm. Biomed. 2013, 84, 209–214. [Google Scholar] [CrossRef]
- Wollein, U.; Bauer, B.; Habernegg, R.; Schramek, N. Potential metal impurities in active pharmaceutical substances and finished medicinal products—A market surveillance study. Eur. J. Pharm. Sci. 2015, 77, 100–105. [Google Scholar] [CrossRef]
- Nunes, T.S.; Muller, C.C.; Balestrin, P.; Muller, A.L.H.; Mesko, M.F.; Mello, P.A.; Muller, E.I. Determination of chlorine and sulfur in high purity flexible graphite using ion chromatography (IC) and inductively coupled plasma optical emission spectrometry (ICP OES) after pyrohydrolysis sample preparation. Anal. Methods 2015, 7, 2129–2134. [Google Scholar] [CrossRef]
- Barin, J.S.; Tischer, B.; Picoloto, R.S.; Antes, F.G.; Silva, F.E.B.; Paula, F.R.; Flores, E.M.M. Determination of toxic elements in tricyclic active pharmaceutical ingredients by ICP-MS: A critical study of digestion methods. J. Anal. At. Spectrom. 2014, 29, 352–358. [Google Scholar] [CrossRef]
- Chahrour, O.; Malone, J.; Collins, M.; Salmon, V.; Greenan, C.; Bombardier, A.; Ma, Z.; Dunwoody, N. Development and validation of an ICP-MS method for the determination of elemental impurities in TP-6076 active pharmaceutical ingredient (API) according to USP. J. Pharm. Biomed. 2017, 145, 84–90. [Google Scholar] [CrossRef]
- Fischer, L.; Zipfel, B.; Koellensperger, G.; Kovac, J.; Bilz, S.; Kunkel, A.; Venzago, C.; Hann, S. Flow injection combined with ICP-MS for accurate high throughput analysis of elemental impurities in pharmaceutical products according to USP. J. Pharm. Biomed. 2014, 95, 121–129. [Google Scholar] [CrossRef]
- Gonzalez, M.H.; Silva, C.S.; Amaral, C.D.B.; Bianchi, S.R.; Oliveira, L.H.B.; Coelho, J.S.; Oliveira, A.; Nogueira, A.R.A. Determination of Elemental Impurities in Acyclovir Ointment and Raw Materials Using Microwave Acid Digestion (MW-AD) and ICP-MS. J. Braz. Chem. Soc. 2017, 28, 98–105. [Google Scholar] [CrossRef]
- Li, G.; Schoneker, D.; Ulman, K.L.; Sturm, J.J.; Thackery, L.M.; Kauffman, J.F. Elemental impurities in pharmaceutical excipients. J. Pharm. Sci. 2015, 104, 4197–4206. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.L.H.; Oliveira, J.S.S.; Mello, P.A.; Muller, E.I.; Flores, E.M.M. Study and determination of elemental impurities by ICP-MS in active pharmaceutical ingredients using single reaction chamber digestion in compliance with USP requirements. Talanta 2015, 136, 161–169. [Google Scholar] [CrossRef]
- Barin, J.S.; Pereira, J.S.; Mello, P.A.; Knorr, C.L.; Moraes, D.P.; Mesko, M.F.; Nóbrega, J.A.; Korn, M.G.; Flores, E.M. Focused microwave-induced combustion for digestion of botanical samples and metals determination by ICP OES and ICP-MS. Talanta 2012, 94, 308–314. [Google Scholar] [CrossRef]
- Hill, S.J. Inductively Coupled Plasma Spectrometry and Its Applications, 2nd ed.; Blackwell Publishing: Oxford, UK, 2007; p. 446. [Google Scholar]
- Grindlay, G.; Mora, J.; Loos-Vollebregt, M.; Vanhaecke, F. A systematic study on the influence of carbon on the behavior of hard-to-ionize elements in inductively coupled plasma–mass spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2013, 86, 42–49. [Google Scholar] [CrossRef]
- Stepan, M.; Musil, P.; Poussel, E.; Mermet, J.M. Matrix-induced shift effects in axially viewed inductively coupled plasma atomic emission spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2001, 56, 443–453. [Google Scholar] [CrossRef]
- Todolí, J.L.; Gras, L.; Hernandis, V.; Mora, J. Elemental matrix effects in ICP-AES. J. Anal. At. Spectrom. 2002, 17, 142–169. [Google Scholar] [CrossRef]
- Wiltsche, H.; Winkler, M.; Tirk, P. Matrix effects of carbon and bromine in inductively coupled plasma optical emission spectrometry. J. Anal. At. Spectrom. 2015, 30, 2223–2234. [Google Scholar] [CrossRef] [Green Version]
- Tu, Q.; Wang, T.; Antonucci, V. High-efficiency sample preparation with dimethylformamide for multi-element determination in pharmaceutical materials by ICP-AES. J. Pharm. Biomed. 2010, 52, 311–315. [Google Scholar] [CrossRef]
- Pinheiro, F.C.; Barros, A.I.; Nobrega, J.A. Microwave-assisted sample preparation of medicines for determination of elemental impurities in compliance with United States Pharmacopeia: How simple can it be? Anal. Chim. Acta 2019, 1065, 1–11. [Google Scholar] [CrossRef]
- Müller, E.I.; Mesko, M.F.; Moraes, D.P.; Korn, M.d.G.A.; Flores, E.M.M. Chapter 4—Wet Digestion Using Microwave Heating. In Microwave-Assisted Sample Preparation for Trace Element Analysis; Flores, E.M.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 99–142. [Google Scholar]
- Bizzi, C.A.; Flores, E.L.M.; Nobrega, J.A.; Oliveira, J.S.S.; Schmidt, L.; Mortari, S.R. Evaluation of a digestion procedure based on the use of diluted nitric acid solutions and H2O2 for the multielement determination of whole milk powder and bovine liver by ICP-based techniques. J. Anal. At. Spectrom. 2014, 29, 332–338. [Google Scholar] [CrossRef]
- Bizzi, C.A.; Nóbrega, J.A.; Barin, J.S. Chapter 6—Diluted Acids in Microwave-Assisted Wet Digestion. In Microwave-Assisted Sample Preparation for Trace Element Analysis; Flores, E.M.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 179–204. [Google Scholar]
- Bizzi, C.A.; Barin, J.S.; Muller, E.I.; Schmidt, L.; Nobrega, J.A.; Flores, E.M.M. Evaluation of oxygen pressurized microwave-assisted digestion of botanical materials using diluted nitric acid. Talanta 2011, 83, 1324–1328. [Google Scholar] [CrossRef]
- Bizzi, C.A.; Nobrega, J.A.; Barin, J.S.; Oliveira, J.S.S.; Schmidt, L.; Mello, P.A.; Flores, E.M.M. Effect of simultaneous cooling on microwave-assisted wet digestion of biological samples with diluted nitric acid and O2 pressure. Anal. Chim. Acta 2014, 837, 16–22. [Google Scholar] [CrossRef]
- Pardinho, R.B.; Dalla Vecchia, P.; Mendes, A.L.G.; Bizzi, C.A.; Mello, P.A.; Duarte, F.A.; Flores, E.M.M. Determination of toxic elements in yerba mate by ICP-MS after diluted acid digestion under O2 pressure. Food Chem. 2018, 263, 37–41. [Google Scholar] [CrossRef]
- Pereira, J.S.F.; Wiltsche, H.; Knapp, G. Chapter 7—Microwave-Assisted Ultraviolet Digestion. In Microwave-Assisted Sample Preparation for Trace Element Analysis; Flores, E.M.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 205–229. [Google Scholar]
- Iop, G.D.; Krzyzaniak, S.R.; Silva, J.S.; Flores, E.M.M.; Costa, A.B.; Mello, P.A. Feasibility of microwave-assisted ultraviolet digestion of polymeric waste electrical and electronic equipment for the determination of bromine and metals (Cd, Cr, Hg, Pb and Sb) by ICP-MS. J. Anal. At. Spectrom. 2017, 32, 1789–1797. [Google Scholar] [CrossRef]
- Hartwig, C.A.; Pereira, R.M.; Rondan, F.S.; Cruz, S.M.; Duarte, F.A.; Flores, E.M.M.; Mesko, M.F. The synergic effect of microwave and ultraviolet radiation for chocolate digestion and further determination of As, Cd, Ni and Pb by ICP-MS. J. Anal. At. Spectrom. 2016, 31, 523–530. [Google Scholar] [CrossRef]
- Picoloto, R.S.; Pereira, R.M.; Costa, V.C.; Hartwig, C.A.; Pereira, C.M.P.; Colepicolo, P.; Duarte, F.A.; Mesko, M.F. Investigating essential and toxic elements in Antarctic macroalgae using a green analytical method. J. Appl. Phycol. 2017, 29, 741–749. [Google Scholar] [CrossRef]
- Mesko, M.F.; Picoloto, R.S.; Ferreira, L.R.; Costa, V.C.; Pereira, C.M.P.; Colepicolo, P.; Muller, E.I.; Flores, E.M.M. Ultraviolet radiation combined with microwave-assisted wet digestion of Antarctic seaweeds for further determination of toxic elements by ICP-MS. J. Anal. At. Spectrom. 2015, 30, 260–266. [Google Scholar] [CrossRef]
- Soares, B.M.; Vieira, A.A.; Lemões, J.S.; Santos, C.M.; Mesko, M.F.; Primel, E.G.; Montes D’Oca, M.G.; Duarte, F.A. Investigation of major and trace element distribution in the extraction-transesterification process of fatty acid methyl esters from microalgae Chlorella sp. Bioresour. Technol. 2012, 110, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.S.; Picoloto, R.S.; Pereira, L.S.; Guimaraes, R.C.; Guarnieri, R.A.; Flores, E.M.M. High-efficiency microwave-assisted digestion combined to in situ ultraviolet radiation for the determination of rare earth elements by ultrasonic nebulization ICPMS in crude oils. Anal. Chem. 2013, 85, 11034–11040. [Google Scholar] [CrossRef]
- Souza, J.P.; Barela, P.S.; Kellermann, K.; Santos, M.F.P.; Moraes, D.P.; Pereira, J.S.F. Microwave-assisted ultraviolet digestion: An efficient method for the digestion of produced water from crude oil extraction and further metal determination. J. Anal. At. Spectrom. 2017, 32, 2439–2446. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Li, G.; Qu, J. Oxidative decomposition of azo dye C.I. Acid Orange 7 (AO7) under microwave electrodeless lamp irradiation in the presence of H2O2. J. Hazard. Mater. 2006, 134, 183–189. [Google Scholar] [CrossRef]
- Nam, K.H.; Isensee, R.; Infantino, G.; Putyera, K.; Wang, X. Microwave-induced combustion for ICP-MS: A generic approach to trace elemental analyses of pharmaceutical products. Spectroscopy 2011, 26, 2–7. [Google Scholar]
- Microwave-Assisted UV Digestion (MUV) Instruction Sheet D19IB018EN-A. Multiwave 3000 Microwave Sample Preparation System; Software Version v1.27-Synt; Anton Paar GmbH: Graz, Austria, 2003; Available online: https://www.anton-paar.com/corp-en/services-support/document-finder/application-reports/microwave-assisted-uv-digestion-of-food-related-liquids/ (accessed on 10 June 2021).
- Würfels, M.; Jackwerth, E.; Stoeppler, M. Residues from biological materials after pressure decomposition with nitric acid: Part 1. Carbon conversion during sample decomposition. Anal. Chim. Acta 1989, 226, 1–16. [Google Scholar] [CrossRef]
- Würfels, M.; Jackwerth, E.; Stoeppler, M. Residues from biological materials after pressure decomposition with nitric acid: Part 2. Identification of the reaction products. Anal. Chim. Acta 1989, 226, 17–30. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | ICP-OES | |
|---|---|---|
| RF power (W) | 1400 | |
| Plasma flow rate (L min−1) | 12.0 | |
| Auxiliary gas flow rate (L min−1) | 1.0 | |
| Nebulizer gas flow rate (L min−1) | 1.00 | |
| Spray chamber | Double path, Scott type | |
| Nebulizer | Cross-flow | |
| Analytes | Emission line, nm | |
| As | 189.042 (I) | |
| Ca | 396.847 (II) | |
| Cd | 214.438 (II) | |
| Co | 230.786 (II) | |
| Fe | 259.941 (II) | |
| Hg | 194.227 (II) | |
| K | 766.491 (I) | |
| Mg | 279.553 (II) | |
| Na | 589.592 (I) | |
| Ni | 231.604 (II) | |
| Pb | 220.353 (II) | |
| V | 292.402 (II) | |
| C | 193.030 (I) | |
| Y | 371.029 (II) | |
| Sample ID | API | API Dose (mg) | Mean Tablet Mass (mg) | N * | Pharmaceutical Class | Excipients |
|---|---|---|---|---|---|---|
| MET | Metformin hydrochlorate | 500 | 600 | 1 | Biguanide | Cornstarch, copolymer of poly(vinyl alcohol) and macrogol, SiO2, povidone, magnesium stearate, sodium starch glycolate, macrogol |
| GLIB | Glibenclamide | 5 | 125 | 4 | Sulfonylurea | Lactose monohydrate, povidone, crospovidone, magnesium stearate |
| SITA | Sitagliptin phosphate | 50 | 210 | 2 | DPP-4 inhibitor | Microcrystalline cellulose, CaHPO4, croscarmellose sodium, sodium starch glycolate, magnesium stearate, sodium stearyl fumarate, poly(vinyl alcohol), macrogol, talc, TiO2, Fe2O3, FeO |
| PIO | Pioglitazone hydrochloride | 45 | 180 | 3 | Thiazolidinedione | Lactose monohydrate, croscarmellose sodium, sodium starch glycolate, hyprolosis, magnesium stearate, H2O |
| CANA | Canagliflozin | 100 | 210 | 2 | SGLT2 inhibitor | Microcrystalline cellulose, anhydrous lactose, croscarmellose sodium, hyprolosis, magnesium stearate, poly(vinyl alcohol), TiO2, macrogol, talc, FeO |
| REPA | Repaglinide | 2 | 100 | 5 | Meglitinide | Microcrystalline cellulose, CaPO4·2H2O, CaCO3, cornstarch, povidone, crospovidone, sodium lauryl sulfate, magnesium stearate, Fe2O3 |
| Sample | Element | ||||
|---|---|---|---|---|---|
| Ca | Fe | K | Mg | Na | |
| CANA a | 27.7 ± 2.0 | 142 ± 1 | 26.1 ± 1.0 | 574 ± 2 | 2964 ± 24 |
| GLIB b | 96.2 ± 3.3 | 3.95 ± 0.90 | 84.0 ± 2.2 | 304 ± 6 | <83.9 |
| MET b | 20.5 ± 0.8 | 2.25 ± 0.29 | 10.1 ± 1.0 | 1376 ± 40 | 1856 ± 140 |
| PIO b | 17.8 ± 0.8 | 3.50 ± 0.29 | 59.1 ± 0.8 | 191 ± 13 | 5129 ± 267 |
| REPA b | <10.8 | 101 ± 4 | 83.4 ± 0.8 | 1407 ± 82 | 4865 ± 113 |
| SITA b | <10.8 | 242 ± 5 | 27.9 ± 1.4 | 1139 ± 75 | 3012 ± 35 |
| Analyte | CRM BCR 60 | CRM DOLT-4 | CRM TORT-2 | |||
|---|---|---|---|---|---|---|
| Obtained Value | Certified Value | Obtained Value | Certified Value | Obtained Value | Certified Value | |
| MAWD | ||||||
| As | 6.87 ± 0.19 | 8.00 * | 9.71 ± 0.53 | 9.66 ± 0.62 | 22.0 ± 0.5 | 21.6 ± 1.8 |
| Cd | 2.19 ± 0.02 | 2.20 ± 0.10 | 24.5 ± 0.2 | 24.3 ± 0.8 | 26.7 ± 0.3 | 26.7 ± 0.6 |
| Co | 3.97 ± 0.09 | 4.00 * | <0.64 | 0.250 * | <0.64 | 0.510 ± 0.090 |
| Hg | <0.77 | 0.340 ± 0.040 | 2.62 ± 0.06 | 2.58 ± 0.22 | <0.77 | 0.270 ± 0.060 |
| Ni | 40.1 ± 2.1 | 40.0 * | 0.992 ± 0.073 | 0.970 ± 0.110 | 2.53 ± 0.06 | 2.50 ± 0.19 |
| Pb | 63.5 ± 1.9 | 63.8 ± 3.2 | <1.22 | 0.160 ± 0.023 | <1.22 | 0.350 ± 0.130 |
| V | 4.75 ± 0.09 | 6.00 * | 0.575 ± 0.039 | 0.600 * | 1.66 ± 0.03 | 1.64 ± 0.19 |
| MAWD-UV | ||||||
| As | 7.08 ± 0.19 | 8.00 * | 9.44 ± 0.57 | 9.66 ± 0.62 | 21.0 ± 0.5 | 21.6 ± 1.8 |
| Cd | 2.21 ± 0.03 | 2.20 ± 0.10 | 25.7 ± 0.3 | 24.3 ± 0.8 | 26.4 ± 0.4 | 26.7 ± 0.6 |
| Co | 3.91 ± 0.07 | 4.00 * | <0.48 | 0.250 * | 0.507 ± 0.014 | 0.510 ± 0.090 |
| Hg | <0.65 | 0.340 ± 0.040 | 2.41 ± 0.07 | 2.58 ± 0.22 | <0.65 | 0.270 ± 0.060 |
| Ni | 38.7 ± 1.1 | 40.0 * | 1.06 ± 0.06 | 0.970 ± 0.110 | 2.49 ± 0.09 | 2.50 ± 0.19 |
| Pb | 64.2 ± 2.5 | 63.8 ± 3.2 | <1.09 | 0.160 ± 0.023 | <1.09 | 0.350 ± 0.130 |
| V | 4.25 ± 0.07 | 6.00 * | 0.549 ± 0.053 | 0.600 * | 1.76 ± 0.05 | 1.64 ± 0.19 |
| Analyte | MAWD | MAWD-UV |
|---|---|---|
| As | 1.57 | 1.80 |
| Cd | 0.06 | 0.07 |
| Co | 0.64 | 0.48 |
| Hg | 0.77 | 0.65 |
| Ni | 0.50 | 0.64 |
| Pb | 1.22 | 1.09 |
| V | 0.10 | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cauduro, V.H.; Henn, A.S.; Picoloto, R.S.; Muller, E.I.; Mesko, M.F.; Flores, E.M.M. Development of Green Methods for the Determination of Elemental Impurities in Commercial Pharmaceutical Tablets. Sustainability 2022, 14, 422. https://doi.org/10.3390/su14010422
Cauduro VH, Henn AS, Picoloto RS, Muller EI, Mesko MF, Flores EMM. Development of Green Methods for the Determination of Elemental Impurities in Commercial Pharmaceutical Tablets. Sustainability. 2022; 14(1):422. https://doi.org/10.3390/su14010422
Chicago/Turabian StyleCauduro, Vitoria Hagemann, Alessandra Schneider Henn, Rochele Sogari Picoloto, Edson Irineu Muller, Marcia Foster Mesko, and Erico Marlon Moraes Flores. 2022. "Development of Green Methods for the Determination of Elemental Impurities in Commercial Pharmaceutical Tablets" Sustainability 14, no. 1: 422. https://doi.org/10.3390/su14010422