
Chemotherapeutic Potential of Saikosaponin D: Experimental Evidence


Abstract
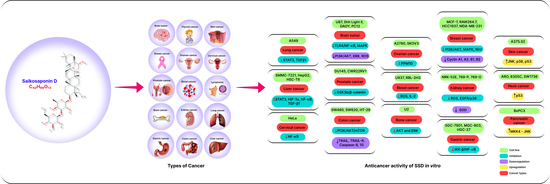
:
1. Introduction
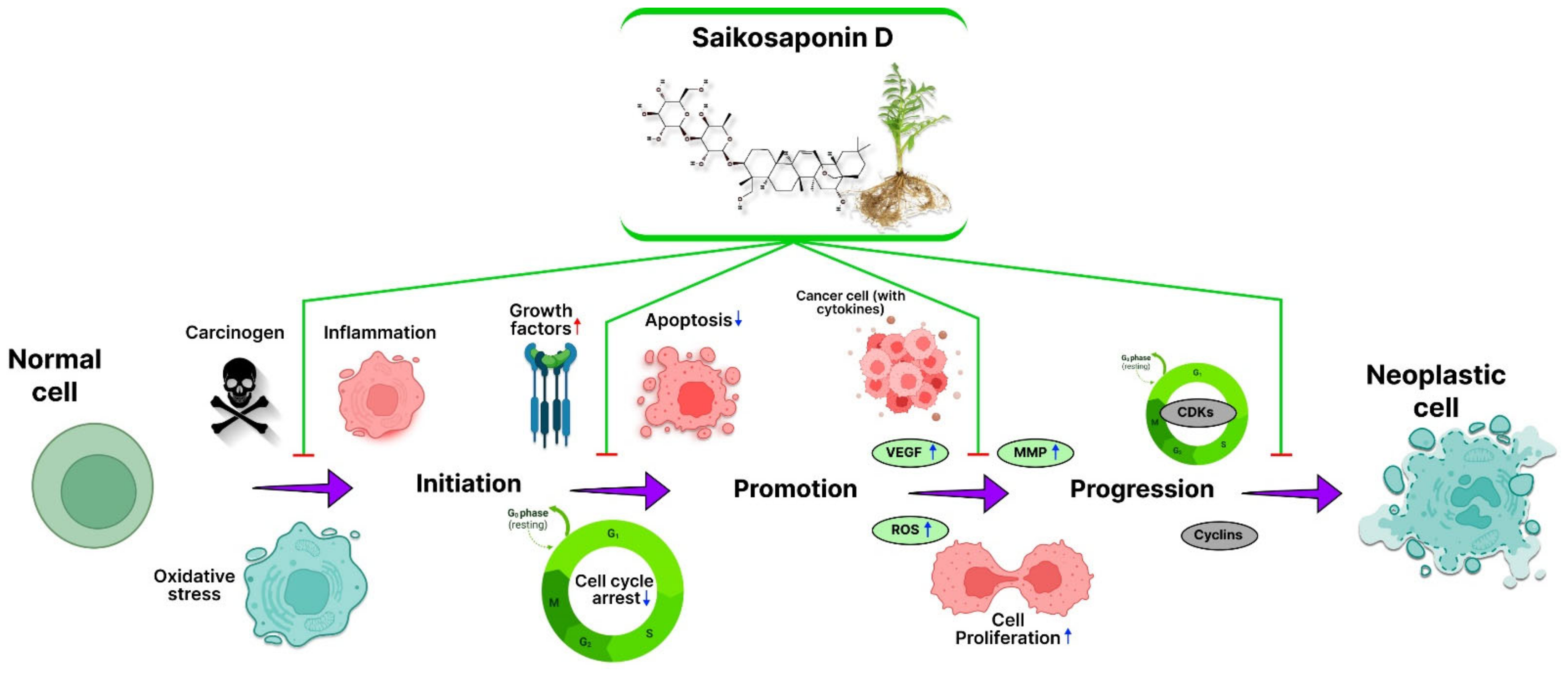
2. SSD in Inhibition of Cancer: Mechanism
2.1. Inflammation
2.2. Reactive Oxygen Species (ROS)
2.3. Angiogenesis
2.4. Apoptosis
2.5. Cell Cycle
2.6. Signal Transducer and Activator of Transcription 3 (STAT3)
3. Anticancer Effects
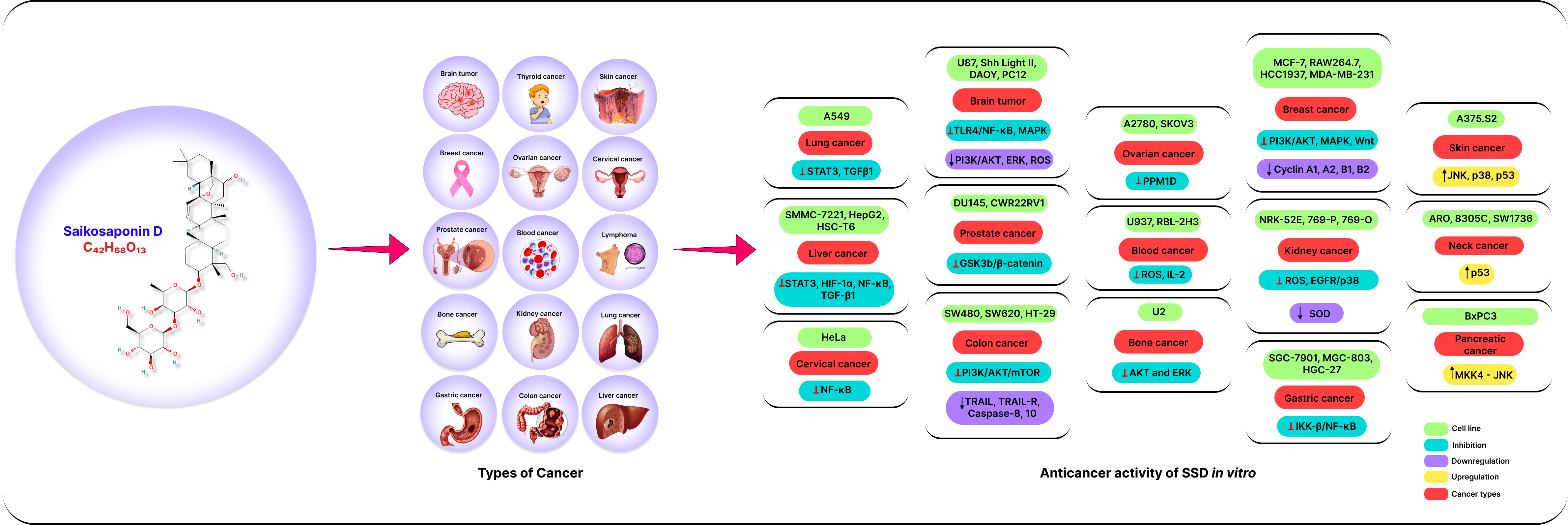
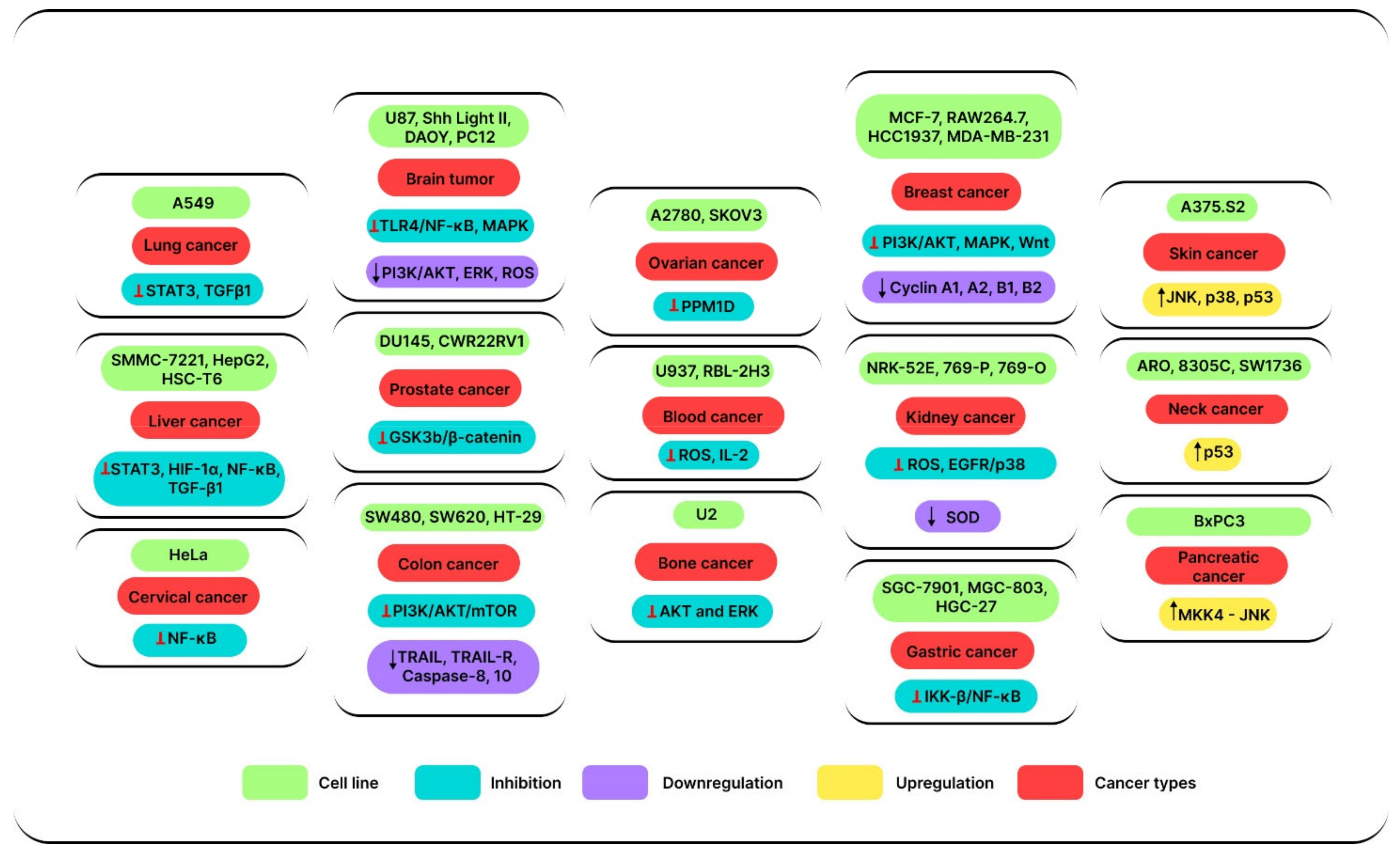
3.1. Anticancer Effects of SSD In Vitro
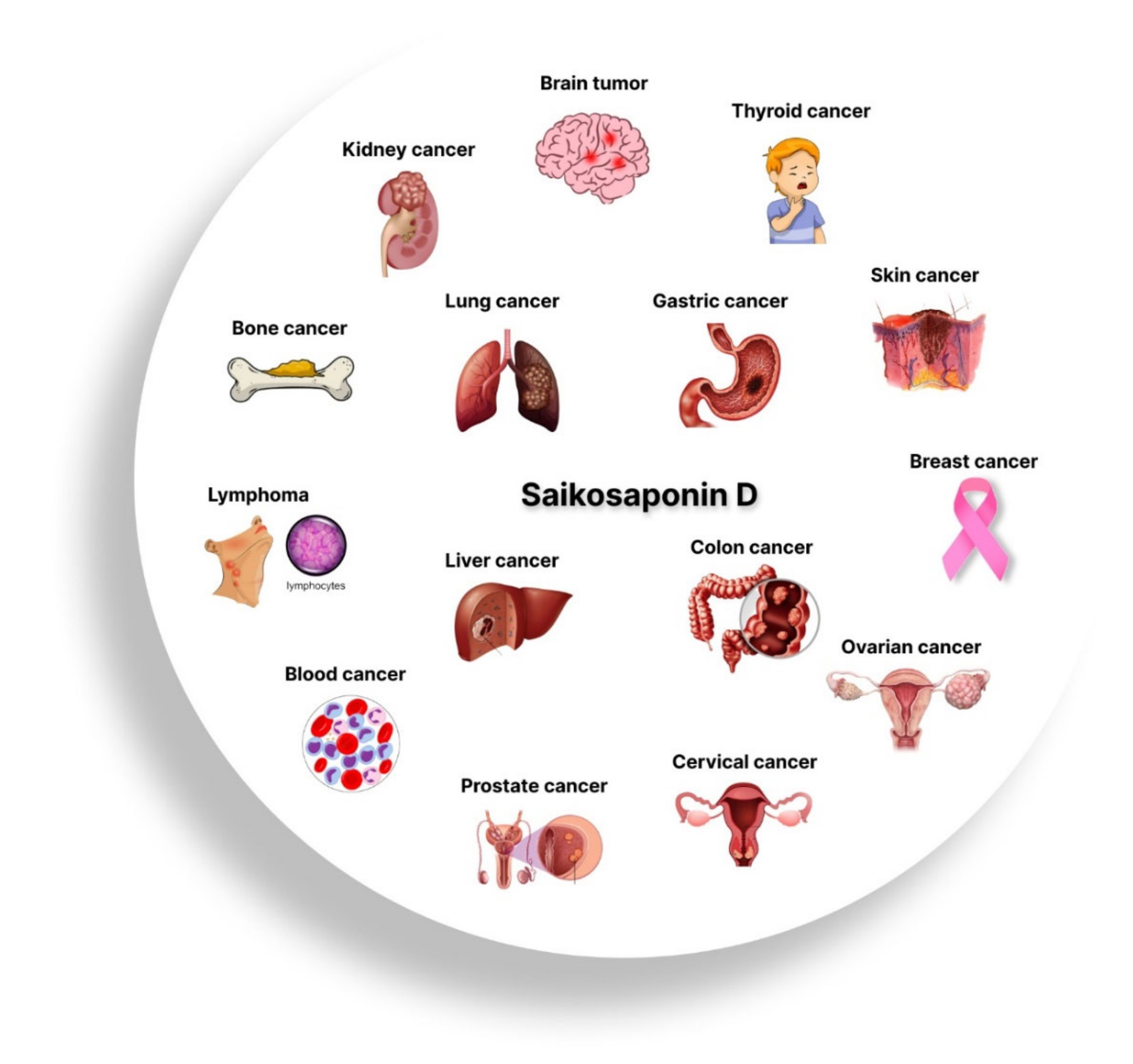
3.1.1. Lung Cancer
3.1.2. Breast Cancer
3.1.3. Liver Cancer
3.1.4. Kidney Cancer
3.1.5. Cervical Cancer
3.1.6. Blood Cancer
3.1.7. Lymphoma
3.1.8. Brain Tumor
3.1.9. Ovarian Cancer
3.1.10. Prostate Cancer
3.1.11. Bone Cancer
3.1.12. Colon Cancer
3.1.13. Thyroid Cancer
3.1.14. Skin Cancer
3.1.15. Stomach Cancer
3.2. Anticancer Effects of SSD In Vivo
3.2.1. Lung Cancer
3.2.2. Breast Cancer
3.2.3. Liver Cancer
3.2.4. Kidney Cancer
3.2.5. Blood Cancer
3.2.6. Brain Tumor
3.2.7. Thyroid Cancer
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Steward, W.P.; Brown, K. Cancer chemoprevention: A rapidly evolving field. Br. J. Cancer 2013, 109, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.; Deep, G.; Agarwal, C.; Agarwal, R. The strategies to control prostate cancer by chemoprevention approaches. Mutat. Res. Mol. Mech. Mutagen. 2014, 760, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxe, G.A.; Major, J.M.; Westerberg, L.; Khandrika, S.; Downs, T.M. Biological Mediators of Effect of Diet and Stress Reduction on Prostate Cancer. Integr. Cancer Ther. 2008, 7, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishayee, A.; Ahmed, S.; Brankov, N.; Perloff, M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front. Biosci. 2011, 16, 980–996. [Google Scholar] [CrossRef] [Green Version]
- Bommareddy, A.; Eggleston, W.; Prelewicz, S.; Antal, A.; Witczak, Z.; McCune, D.F.; VanWert, A.L. Chemoprevention of prostate cancer by major dietary phytochemicals. Anticancer Res. 2013, 33, 4163–4174. [Google Scholar] [PubMed]
- Wang, P.; Wang, B.; Chung, S.; Wu, Y.; Henning, S.M.; Vadgama, J.V. Increased chemopreventive effect by combining arctigenin, green tea polyphenol and curcumin in prostate and breast cancer cells. RSC Adv. 2014, 4, 35242–35250. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Lin, S.-R.; Fu, Y.-S.; Tsai, M.-J.; Cheng, H.; Weng, C.-F. Natural Compounds from Herbs that can Potentially Execute as Autophagy Inducers for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1412. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Yao, R.; Chen, X.; Yang, X.; Li, Z. Advances in studies on classification of Bupleurum. China J. Chin. Mater. Medica 2012, 37, 1523–1526. [Google Scholar]
- Ashour, M.L.; Wink, M. Genus Bupleurum: A review of its phytochemistry, pharmacology and modes of action. J. Pharm. Pharmacol. 2011, 63, 305–321. [Google Scholar] [CrossRef]
- Jiang, H.; Yang, L.; Hou, A.; Zhang, J.; Wang, S.; Man, W.; Zheng, S.; Yu, H.; Wang, X.; Yang, B.; et al. Botany, traditional uses, phytochemistry, analytical methods, processing, pharmacology and pharmacokinetics of Bupleuri Radix: A systematic review. Biomed. Pharmacother. 2020, 131, 110679. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhu, Z.; Li, A.; Hu, Z.; Yang, N.; Ying, Z.; Wang, C.; Yin, S.; Cheng, S. Recent Advances in Biotransformation of Saponins. Molecules 2019, 24, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Perumal, E. Potential Role of Marine Bioactive Compounds Targeting Signaling Pathways in Cancer: A Review. Eur. J. Pharmacol. 2022, 936, 175330. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Huang, N.; Liu, R.; Sun, R. A comprehensive review and perspectives on pharmacology and toxicology of saikosaponins. Phytomedicine 2018, 50, 73–87. [Google Scholar] [CrossRef]
- Lin, H.-J.; Kao, S.-T.; Siao, Y.; Yeh, C.-C. The Chinese medicine Sini-San inhibits HBx-induced migration and invasiveness of human hepatocellular carcinoma cells. BMC Complement. Altern. Med. 2015, 15, 348. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Li, K.; Long, S.; Kong, C.; Zhu, S. Potential Molecular Mechanisms of Chaihu-Shugan-San in Treatment of Breast Cancer Based on Network Pharmacology. Evid.-Based Complement. Altern. Med. 2020, 2020, 3670309. [Google Scholar] [CrossRef]
- Li, D.-Q.; Wu, J.; Liu, L.-Y.; Wu, Y.-Y.; Li, L.-Z.; Huang, X.-X.; Liu, Q.-B.; Yang, J.-Y.; Song, S.-J.; Wu, C.-F. Cytotoxic triterpenoid glycosides (saikosaponins) from the roots of Bupleurum chinense. Bioorganic Med. Chem. Lett. 2015, 25, 3887–3892. [Google Scholar] [CrossRef]
- Yuan, B.; Yang, R.; Ma, Y.; Zhou, S.; Zhang, X.; Liu, Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm. Biol. 2017, 55, 620–635. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.C.-S.; Lai, Y.-C.; Lin, C.-L.; Tzeng, W.-S.; Yen, F.-L. Inclusion complex of saikosaponin-d with hydroxypropyl-β-cyclodextrin: Improved physicochemical properties and anti-skin cancer activity. Phytomedicine 2019, 57, 174–182. [Google Scholar] [CrossRef]
- Du, J.-R.; Long, F.-Y.; Chen, C. Research Progress on Natural Triterpenoid Saponins in the Chemoprevention and Chemo-therapy of Cancer. Enzym. 2014, 36, 95–130. [Google Scholar]
- Kim, J.-K.; Oh, S.-m.; Kwon, H.-S.; Oh, Y.-S.; Lim, S.S.; Shin, H.-K. Anti-inflammatory effect of roasted licorice extracts on lipopolysaccharide-induced inflammatory responses in murine macrophages. Biochem. Biophys. Res. Commun. 2006, 345, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.M.; Zhou, H.Y.; Guo, L.Y.; Kim, J.A.; Lee, S.H.; Merfort, I.; Kang, S.S.; Kim, H.S.; Kim, S.; Kim, Y.S. Anti-inflammatory effects of glycyrol isolated from Glycyrrhiza uralensis in LPS-stimulated RAW264.7 macrophages. Int. Immunopharmacol. 2008, 8, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-N.; Yuan, Z.-G.; Zhang, X.-L.; Yan, R.; Zhao, Y.-Q.; Liao, M.; Chen, J.-X. Saikosaponin a and its epimer saikosaponin d exhibit anti-inflammatory activity by suppressing activation of NF-κB signaling pathway. Int. Immunopharmacol. 2012, 14, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Pan, Y.-W.; Wang, S.-Q.; Li, X.-Z.; Huang, F.; Ma, S.-P. Saikosaponin-d attenuated lipopolysaccharide-induced depressive-like behaviors via inhibiting microglia activation and neuroinflammation. Int. Immunopharmacol. 2020, 80, 106181. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Westerblad, H.; Allen, D.G. Emerging Roles of ROS/RNS in Muscle Function and Fatigue. Antioxid. Redox Signal. 2011, 15, 2487–2499. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Wu, S.; Wang, Q.; Shi, Y.; Liu, G.; Zhi, J.; Wang, F. Saikosaponin-D Reduces H2O2-Induced PC12 Cell Apoptosis by Removing ROS and Blocking MAPK-Dependent Oxidative Damage. Cell. Mol. Neurobiol. 2016, 36, 1365–1375. [Google Scholar] [CrossRef]
- Lim, H.J.; Park, M.N.; Kim, C.; Kang, B.; Song, H.-S.; Lee, H.; Kim, S.-H.; Shim, B.-S.; Kim, B. MiR-657/ATF2 Signaling Pathway Has a Critical Role in Spatholobus suberectus Dunn Extract-Induced Apoptosis in U266 and U937 Cells. Cancers 2019, 11, 150. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Dang, S.; Cheng, Y.; Zhang, X.; Li, M.; Li, Y.; Li, S. Effects of Saikosaponin-D on syndecan-2, matrix metalloproteinases and tissue inhibitor of metalloproteinases-2 in rats with hepatocellular carcinoma. J. Tradit. Chin. Med. 2012, 32, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, V.K.W.; Zhang, M.M.; Zhou, H.; Lam, K.Y.C.; Chan, P.L.; Law, C.K.M.; Yue, P.Y.K.; Liu, L. Saikosaponin-d Enhances the Anticancer Potency of TNF-αvia Overcoming Its Undesirable Response of Activating NF-Kappa B Signalling in Cancer Cells. Evidence-Based Complement. Altern. Med. 2013, 2013, 745295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Bostancıklıoğlu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef]
- Fu, R.; Zhang, L.; Li, Y.; Li, B.; Ming, Y.; Li, Z.; Xing, H.; Chen, J. Saikosaponin D inhibits autophagosome-lysosome fusion and induces autophagy-independent apoptosis in MDA-MB-231 breast cancer cells. Mol. Med. Rep. 2020, 22, 1026–1034. [Google Scholar] [CrossRef]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.-F.; Dai, Z.-J.; Wang, X.-J.; Bai, M.-H.; Lin, S.; Ma, H.-B.; Wang, Y.-L.; Song, L.-Q.; Ma, X.-L.; Zan, Y.; et al. Saikosaponin-d increases the radiosensitivity of smmc-7721 hepatocellular carcinoma cells by adjusting the g0/g1 and g2/m checkpoints of the cell cycle. BMC Complement. Altern. Med. 2013, 13, 263. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; YiLan, J.; YongMin, L.; XiaoNing, T.; Yuan, L.; Ji, L. Effects of Saikosaponin D on the Proliferation, Cell Cycle and the Expressions of Cyclins in Breast Cancer MDA-MB-231 Cells. Chin. J. Integr. Tradit. West. Med. 2019, 39, 572–576. [Google Scholar]
- Sehgal, P.B. Paradigm shifts in the cell biology of STAT signaling. Semin. Cell Dev. Biol. 2008, 19, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tanaka, N.; Sun, L.; Guo, B.; Kim, J.-H.; Krausz, K.W.; Fang, Z.; Jiang, C.; Yang, J.; Gonzalez, F.J. Saikosaponin d protects against acetaminophen-induced hepatotoxicity by inhibiting NF-κB and STAT3 signaling. Chem. Interact. 2014, 223, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; He, S.; Chen, H.; Zhu, X.; McGowan, E. A novel anti-tumorigenic mechanism by herbal extract saikosaponin-d through p-STAT3/C/EBPβ signaling suppression of COX-2 in liver cancer. Ann. Oncol. 2019, 30, iv71. [Google Scholar] [CrossRef]
- Manoharan, S.; Balakrishnan, A.; Hemamalini, V.; Perumal, E. Screening of potent STAT3-SH2 domain inhibitors from JAK/STAT compound library through molecular dynamics simulation. Mol. Divers. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Ren, M.; McGowan, E.; Li, Y.; Zhu, X.; Lu, X.; Zhu, Z.; Lin, Y.; He, S. Saikosaponin-d Suppresses COX2 Through p-STAT3/C/EBPβ Signaling Pathway in Liver Cancer: A Novel Mechanism of Action. Front. Pharmacol. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.-L.; Kuo, P.-L.; Lin, C.-C. The proliferative inhibition and apoptotic mechanism of Saikosaponin D in human non-small cell lung cancer A549 cells. Life Sci. 2004, 75, 1231–1242. [Google Scholar] [CrossRef]
- Wu, S.; Chen, W.; Liu, K.; Ren, F.; Zheng, D.; Xu, F.; Wu, H. Saikosaponin D inhibits proliferation and induces apoptosis of non-small cell lung cancer cells by inhibiting the STAT3 pathway. J. Int. Med. Res. 2020, 48, 0300060520937163. [Google Scholar] [CrossRef]
- Tang, J.-C.; Long, F.; Zhao, J.; Hang, J.; Ren, Y.-G.; Chen, J.-Y.; Mu, B. The Effects and Mechanisms by which Saikosaponin-D Enhances the Sensitivity of Human Non-small Cell Lung Cancer Cells to Gefitinib. J. Cancer 2019, 10, 6666–6672. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, X.-L.; Yang, L.; Shi, F.; Gao, L.-B.; Zhong, Y.-J.; Sun, H.; He, F.; Lin, Y.; Wang, X. Reactive oxygen species-mediated apoptosis contributes to chemosensitization effect of saikosaponins on cisplatin-induced cytotoxicity in cancer cells. J. Exp. Clin. Cancer Res. 2010, 29, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liu, C.; Zhao, R.; Zhao, P.; Wu, J.; Zhou, N.; Ying, M. Synergetic and Antagonistic Molecular Effects Mediated by the Feedback Loop of p53 and JNK between Saikosaponin D and SP600125 on Lung Cancer A549 Cells. Mol. Pharm. 2018, 15, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Wang, Z.; Zhou, J. Anti-Fibrosis Effect of Saikosaponin D on Pulmonary Fibrosis in Vivo and in Vitro via Suppressing Alveolar Epithelial Cell Apoptosis and Epithelium-Mesenchymal Transformation. Int. J. Clin. Exp. Med. 2017, 10, 4417–4425. [Google Scholar]
- Sun, K.; Yu, W.; Ji, B.; Chen, C.; Yang, H.; Du, Y.; Song, M.; Cai, H.; Yan, F.; Su, R. Saikosaponin D loaded macrophage membrane-biomimetic nanoparticles target angiogenic signaling for breast cancer therapy. Appl. Mater. Today 2020, 18, 100505. [Google Scholar] [CrossRef]
- Wang, J.; Qi, H.; Zhang, X.; Si, W.; Xu, F.; Hou, T.; Zhou, H.; Wang, A.; Li, G.; Liu, Y.; et al. Saikosaponin D from Radix Bupleuri suppresses triple-negative breast cancer cell growth by targeting β-catenin signaling. Biomed. Pharmacother. 2018, 108, 724–733. [Google Scholar] [CrossRef]
- Li, D.; Liu, D.; Yue, D.; Gao, P.; Du, C.; Liu, X.; Zhang, L. Network pharmacology and RNA sequencing studies on triterpenoid saponins from Bupleurum chinense for the treatment of breast cancer. RSC Adv. 2019, 9, 41088–41098. [Google Scholar] [CrossRef] [Green Version]
- Kars, M.D.; Kars, G.; Gunduz, U. Paclitaxel Resistance in MCF-7/Pac Cell Line Is Reversed Successfully by Saikosaponin A and Saikosaponin D. UHOD-Uluslar. Hematol.-Onkol. Derg. 2013, 23, 227–232. [Google Scholar] [CrossRef]
- Li, C.; Xue, H.-G.; Feng, L.-J.; Wang, M.-L.; Wang, P.; Gai, X.-D. The Effect of Saikosaponin D on Doxorubicin Pharmacokinetics and Its MDR Reversal in MCF-7/Adr Cell Xenografts. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4437–4445. [Google Scholar]
- Li, C.; Guan, X.; Xue, H.; Wang, P.; Wang, M.; Gai, X. Reversal of P-glycoprotein-mediated multidrug resistance is induced by saikosaponin D in breast cancer MCF-7/adriamycin cells. Pathol.-Res. Pract. 2017, 213, 848–853. [Google Scholar] [CrossRef]
- He, S.; Lu, G.; Hou, H.; Zhao, Z.; Zhu, Z.; Lu, X.; Chen, J.; Wang, Z. Saikosaponin-d suppresses the expression of cyclooxygenase-2 through the phospho-signal transducer and activator of transcription 3/hypoxia-inducible factor-1α pathway in hepatocellular carcinoma cells. Mol. Med. Rep. 2014, 10, 2556–2562. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Min, W.; Lin, S.; Song, L.; Yang, P.; Ma, Q.; Guo, J. Saikosaponin-d increases radiation-induced apoptosis of hepatoma cells by promoting autophagy via inhibiting mTOR phosphorylation. Int. J. Med. Sci. 2021, 18, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Hou, X.; Zhang, Q.; Li, R.; Xu, L.; Chen, Y.; Tian, Y.; Sun, R.; Zhang, Z.; Xu, F. Untargeted Metabolomics Study of the In Vitro Anti-Hepatoma Effect of Saikosaponin d in Combination with NRP-1 Knockdown. Molecules 2019, 24, 1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.-L.; Kuo, P.-L.; Chiang, L.-C.; Lin, C.-C. Involvement of p53, nuclear factor κB and Fas/Fas ligand in induction of apoptosis and cell cycle arrest by saikosaponin d in human hepatoma cell lines. Cancer Lett. 2004, 213, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Duan, S.; Ding, M.; Zheng, Q.; Fan, G.; Li, X.; Li, Y.; Liu, C.; Sun, R.; Liu, R. Saikosaponin D attenuates metabolic associated fatty liver disease by coordinately tuning PPARα and INSIG/SREBP1c pathway. Phytomedicine 2022, 103, 154219. [Google Scholar] [CrossRef]
- Zou, C.; Hu, X.; Wu, Y.; Zhao, L.; Liang, R.; Cao, D.; Zhou, M.; Lin, L. Protective role of Saikosaponin d in d-galactosamine and lipopolysaccharide-induced liver injury in hybrid grouper (Epinephelus lanceolatus♂ × Epinephelus fuscoguttatus♀). Aquaculture 2022, 548, 737601. [Google Scholar] [CrossRef]
- Chou, C.; Pan, S.; Teng, C.; Guh, J. Pharmacological evaluation of several major ingredients of Chinese herbal medicines in human hepatoma Hep3B cells. Eur. J. Pharm. Sci. 2003, 19, 403–412. [Google Scholar] [CrossRef]
- Tian, Y.-D.; Lin, S.; Yang, P.-T.; Bai, M.-H.; Jin, Y.-Y.; Min, W.-L.; Ma, H.-B.; Wang, B.-F. Saikosaponin-d Increases the Radiosensitivity of Hepatoma Cells by Adjusting Cell Autophagy. J. Cancer 2019, 10, 4947–4953. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-Y.; Jiang, Z.-M.; Ma, X.-F.; Li, Y.; Liu, X.-Z.; Li, L.-L.; Wu, W.-H.; Wang, T. Saikosaponin-d Inhibits the Hepatoma Cells and Enhances Chemosensitivity Through SENP5-Dependent Inhibition of Gli1 SUMOylation Under Hypoxia. Front. Pharmacol. 2019, 10, 1039. [Google Scholar] [CrossRef]
- Chiang, L.-C.; Ng, L.T.; Liu, L.-T.; Shieh, D.-E.; Lin, C.-C. Cytotoxicity and Anti-Hepatitis B Virus Activities of Saikosaponins from Bupleurum Species. Planta Med. 2003, 69, 705–709. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, F.; Kong, D.; Zhu, X.; Chen, W.; Wang, A.; Zheng, S. Saikosaponin D disrupts platelet-derived growth factor-β receptor/p38 pathway leading to mitochondrial apoptosis in human LO2 hepatocyte cells: A potential mechanism of hepatotoxicity. Chem. Interactions 2013, 206, 76–82. [Google Scholar] [CrossRef]
- Xiao, Y.; Ren, M.; Lu, G.; Zhao, Y.; Zhang, D.; Liu, Y.; Lu, X.; He, S. The Effects of Saikosaponin-d on the Expression of Human Hepatocellular Carcinoma Cell BECN1 and Autophagic Function. J. Xi’an Jiaotong Univ. (Med. Sci.) 2017, 127–130. [Google Scholar] [CrossRef]
- Zhou, H. A Brief History of Bupleuri Radix Research; Heilongjiang University of Chinese Medicine Press: Harbin, China, 2003. [Google Scholar]
- Kang, L.; Yang, F.; Zhang, X.; Zhao, J.; Liu, Y.; Zhao, H.; Hu, Z.; Liu, B.; He, C. Saikosaponin-D Prevents Acute Renal Injury via Inhibition of NLRP3 Inflammasome By SIRT1. Pharm. Chem. J. 2022, 55, 1169–1176. [Google Scholar] [CrossRef]
- Zhang, B.-Z.; Guo, X.-T.; Chen, J.-W.; Zhao, Y.; Cong, X.; Jiang, Z.-L.; Cao, R.-F.; Cui, K.; Gao, S.-S.; Tian, W.-R. Saikosaponin-D Attenuates Heat Stress-Induced Oxidative Damage in LLC-PK1Cells by Increasing the Expression of Anti-Oxidant Enzymes and HSP72. Am. J. Chin. Med. 2014, 42, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J.; et al. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKKβ–AMPK–mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 2018, 449, 219–226. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Bao, J.; Liu, J.; Ji, Z. Saikosaponin-d protects renal tubular epithelial cell against high glucose induced injury through modulation of SIRT3. Int. J. Clin. Exp. Med. 2015, 8, 6472–6481. [Google Scholar]
- Ma, X.; Dang, C.; Kang, H.; Dai, Z.; Lin, S.; Guan, H.; Liu, X.; Wang, X.; Hui, W. Saikosaponin-D reduces cisplatin-induced nephrotoxicity by repressing ROS-mediated activation of MAPK and NF-κB signalling pathways. Int. Immunopharmacol. 2015, 28, 399–408. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, H.; Ou, Y.; Jiang, Y.; Zhong, D.; Qi, H.; Dang, Q. Saikosaponin-d suppresses cell growth in renal cell carcinoma through EGFR/p38 signaling pathway. Neoplasma 2017, 64, 518–525. [Google Scholar] [CrossRef]
- Sun, K.; Du, Y.; Hou, Y.; Zhao, M.; Li, J.; Du, Y.; Zhang, L.; Chen, C.; Yang, H.; Yan, F.; et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m6A signaling. Theranostics 2021, 11, 5831–5846. [Google Scholar] [CrossRef]
- Bu, S.; Xu, J.; Sun, J. Effect of Saikosaponin-d on up-regulating GR mRNA expression and inhibiting cell growth in human leukemia cells. Zhongguo Zhong Xi Yi Jie He Za Zhi 2000, 20, 350–352. [Google Scholar]
- Qi, X.; Fan, M.; Huang, N.; Zhang, X.; Liu, J.; Li, X.; Sun, R. Saikosaponin d contributed to cancer chemotherapy induced neutropenia therapy by promoting neutrophil differentiation via activation CBL-dependent ERK pathway. Pharmacol. Res. 2020, 160, 105149. [Google Scholar] [CrossRef]
- Jang, M.-J.; Kim, Y.S.; Bae, E.Y.; Oh, T.-S.; Choi, H.-J.; Lee, J.-H.; Oh, H.-M.; Lee, S.W. Saikosaponin D Isolated from Bupleurum falcatum Inhibits Selectin-Mediated Cell Adhesion. Molecules 2014, 19, 20340–20349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cai, T.; Zhang, W.; Zhu, W.; Lv, S. Effects of Saikosaponin D on apoptosis in human U87 glioblastoma cells. Mol. Med. Rep. 2017, 16, 1459–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-J.; Chen, I.-L.; Horng, L.-Y.; Wu, R.-T. Induction of differentiation in rat C6 glioma cells with Saikosaponins. Phytotherapy Res. 2002, 16, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.; Yang, J.; Huang, W.; Liu, J.; Tan, W.; Xin, H. Saikosaponin B1 and Saikosaponin D inhibit tumor growth in medulloblastoma allograft mice via inhibiting the Hedgehog signaling pathway. J. Nat. Med. 2022, 76, 584–593. [Google Scholar] [CrossRef]
- Kodama, Y.; Xiaochuan, L.; Tsuchiya, C.; Ohizumi, Y.; Yoshida, M.; Nakahata, N. Dual Effect of Saikogenin D: In Vitro Inhibition of Prostaglandin E2Production and Elevation of Intracellular Free Ca2+Concentration in C6 Rat Glioma Cells. Planta Med. 2003, 69, 765–767. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Jiang, Y.-M.; Liu, Y.-M.; Guo, Z.; Shen, S.-N.; Liu, X.-M.; Pan, R.-L. Saikosaponin D acts against corticosterone-induced apoptosis via regulation of mitochondrial GR translocation and a GR-dependent pathway. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 53, 80–89. [Google Scholar] [CrossRef]
- Tsuyoshi, H.; Wong, V.K.W.; Han, Y.; Orisaka, M.; Yoshida, Y.; Tsang, B.K. Saikosaponin-d, a calcium mobilizing agent, sensitizes chemoresistant ovarian cancer cells to cisplatin-induced apoptosis by facilitating mitochondrial fission and G2/M arrest. Oncotarget 2017, 8, 99825–99840. [Google Scholar] [CrossRef]
- Yao, M.; Yang, J.; Cao, L.; Zhang, L.; Qu, S.; Gao, H. Saikosaponin-d inhibits proliferation of DU145 human prostate cancer cells by inducing apoptosis and arresting the cell cycle at G0/G1 phase. Mol. Med. Rep. 2014, 10, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Zhong, D.; Zhang, H.-J.; Jiang, Y.-D.; Wu, P.; Qi, H.; Cai, C.; Zheng, S.-B.; Dang, Q. Saikosaponin-d: A potential chemotherapeutics in castration resistant prostate cancer by suppressing cancer metastases and cancer stem cell phenotypes. Biochem. Biophys. Res. Commun. 2016, 474, 722–729. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J.; Sun, Z.; Sun, C.; Yu, Z.; Guo, X. Saikosaponin D inhibits proliferation of human osteosarcoma cells via the p53 signaling pathway. Exp. Ther. Med. 2018, 17, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Zhao, P.; Yu, X.; Cao, S.; Zhang, B.; Dai, M. Use of Saikosaponin D and JNK inhibitor SP600125, alone or in combination, inhibits malignant properties of human osteosarcoma U2 cells. Am. J. Transl. Res. 2019, 11, 2070–2080. [Google Scholar] [PubMed]
- Zhang, X.; Liu, Z.; Chen, S.; Li, H.; Dong, L.; Fu, X. A new discovery: Total Bupleurum saponin extracts can inhibit the proliferation and induce apoptosis of colon cancer cells by regulating the PI3K/Akt/mTOR pathway. J. Ethnopharmacol. 2022, 283, 114742. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Sunlu, H.; Yang, J.; Wang, L. Effects of Saikosaponin D on apoptosis genes expression profile of the colon cancer cells HT-29. Afr. J. Pharm. Pharmacol. 2013, 7, 1640–1644. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.K.W.; Zhou, H.; Cheung, S.S.F.; Li, T.; Liu, L. Mechanistic study of saikosaponin-d (Ssd) on suppression of murine T lymphocyte activation. J. Cell. Biochem. 2009, 107, 303–315. [Google Scholar] [CrossRef]
- Hao, Y.; Piao, X.; Piao, X. Saikosaponin-d inhibits β-conglycinin induced activation of rat basophilic leukemia-2H3 cells. Int. Immunopharmacol. 2012, 13, 257–263. [Google Scholar] [CrossRef]
- Liu, R.-Y.; Li, J.-P. Saikosaponin-d Inhibits Proliferation of Human Undifferentiated Thyroid Carcinoma Cells through Induc-tion of Apoptosis and Cell Cycle Arrest. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2435–2443. [Google Scholar]
- Hu, S.C.-S.; Lee, I.-T.; Yen, M.-H.; Lin, C.-C.; Lee, C.-W.; Yen, F.-L. Anti-melanoma activity of Bupleurum chinense, Bupleurum kaoi and nanoparticle formulation of their major bioactive compound saikosaponin-d. J. Ethnopharmacol. 2016, 179, 432–442. [Google Scholar] [CrossRef]
- Lai, M.; Ge, Y.; Chen, M.; Sun, S.; Chen, J.; Cheng, R. Saikosaponin D Inhibits Proliferation and Promotes Apoptosis Through Activation of MKK4–JNK Signaling Pathway in Pancreatic Cancer Cells. OncoTargets Ther. 2020, 13, 9465–9479. [Google Scholar] [CrossRef]
- Hu, J.; Li, P.; Shi, B.; Tie, J. Effects and Mechanisms of Saikosaponin D Improving the Sensitivity of Human Gastric Cancer Cells to Cisplatin. ACS Omega 2021, 6, 18745–18755. [Google Scholar] [CrossRef]
- Wang, H.-W.; Liu, M.; Zhong, T.-D.; Fang, X.-M. Saikosaponin-d attenuates ventilator-induced lung injury in rats. Int. J. Clin. Exp. Med. 2015, 8, 15137. [Google Scholar]
- Wang, B.-F.; Wang, X.-J.; Kang, H.-F.; Bai, M.-H.; Guan, H.-T.; Wang, Z.-W.; Zan, Y.; Song, L.-Q.; Min, W.-L.; Lin, S.; et al. Saikosaponin-D Enhances Radiosensitivity of Hepatoma Cells under Hypoxic Conditions by Inhibiting Hypoxia-Inducible Factor-1a. Cell. Physiol. Biochem. 2014, 33, 37–51. [Google Scholar] [CrossRef] [PubMed]
- He, S.-X.; Lu, X.-L.; Ren, M.-D.; Wang, Y.-L.; Zhang, Y.-X.; Liu, E.-Q. Chemopreventive effect of saikosaponin-d on diethylinitrosamine-induced hepatocarcinogenesis: Involvement of CCAAT/enhancer binding protein�β and cyclooxygenase-2. Mol. Med. Rep. 2011, 5, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, S.-S.; Wang, B.-F.; Cheng, Y.-A.; Song, P.; Liu, Z.-G.; Li, Z.-F. Inhibitory effects of saikosaponin-d on CCl4-induced hepatic fibrogenesis in rats. World J. Gastroenterol. 2007, 13, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, R.; Zhang, L.; Jiang, Z. The emerging role of AMP-activated protein kinase in cholestatic liver diseases. Pharmacol. Res. 2017, 125, 105–113. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Song, T.; Hu, Y.-N.; Lu, X.-L.; Zhang, Y.-X.; Liu, E.-Q.; He, S.-X. Effects of Saikosaponins-d on Ang-2 and VEGF Expressions in Experimental Hepatocarcinoma in Rats. J. Xi’an Jiaotong Univ. (Med. Sci.) 2013, 35, 664–668. [Google Scholar]
- Li, P.; Gong, Y.; Zu, N.; Li, Y.; Wang, B.; Shimizu, F. Therapeutic Mechanism of Saikosaponin-d in Anti-Thy1 mAb 1-22-3-Induced Rat Model of Glomerulonephritis. Nephron 2005, 101, e111–e118. [Google Scholar] [CrossRef]
- Yao, T.; Zhang, L.; Fu, Y.; Yao, L.; Zhou, C.; Chen, G. Saikosaponin-d Alleviates Renal Inflammation and Cell Apoptosis in a Mouse Model of Sepsis via TCF7/FOSL1/Matrix Metalloproteinase 9 Inhibition. Mol. Cell. Biol. 2021, 41, e00332-21. [Google Scholar] [CrossRef]
- Wu, K. Antitumor Activity and Mechanism of Saikosaponin D Combined with Oxaliplatin on A549 Cells-Bearing Nude Mice. J. China Pharm. Univ. 2015, 355–358. Available online: https://pesquisa.bvsalud.org/portal/resource/pt/wpr-811958?lang=en (accessed on 5 November 2022).
- Zhou, L.; Huang, J.; Zhang, D.; Zhao, Y. Cognitive improvements and reduction in amyloid plaque deposition by saikosaponin D treatment in a murine model of Alzheimer’s disease. Exp. Ther. Med. 2020, 20, 1082–1090. [Google Scholar] [CrossRef]
- Kim, P.K.; Park, S.-Y.; Koty, P.P.; Hua, Y.; Luketich, J.D.; Billiar, T.R. Fas-associating death domain protein overexpression induces apoptosis in lung cancer cells. J. Thorac. Cardiovasc. Surg. 2003, 125, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Kalimutho, M.; Nones, K.; Srihari, S.; Duijf, P.H.; Waddell, N.; Khanna, K.K. Patterns of Genomic Instability in Breast Cancer. Trends Pharmacol. Sci. 2019, 40, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Pellacani, D.; Tan, S.; Lefort, S.; Eaves, C.J. Transcriptional regulation of normal human mammary cell heterogeneity and its perturbation in breast cancer. EMBO J. 2019, 38, e100330. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Kim, G.C.; Kweon, S.; Lee, H.; Choi, J.U.; Mahmud, F.; Chang, H.W.; Kim, J.W.; Son, W.-C.; Kim, S.Y.; et al. Metronomic oral doxorubicin in combination of Chk1 inhibitor MK-8776 for p53-deficient breast cancer treatment. Biomaterials 2018, 182, 35–43. [Google Scholar] [CrossRef]
- Bowerman, C.J.; Byrne, J.D.; Chu, K.S.; Schorzman, A.N.; Keeler, A.W.; Sherwood, C.A.; Perry, J.L.; Luft, J.C.; Darr, D.B.; Deal, A.M.; et al. Docetaxel-Loaded PLGA Nanoparticles Improve Efficacy in Taxane-Resistant Triple-Negative Breast Cancer. Nano Lett. 2017, 17, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Cosmai, L.; Leibovich, B.C.; Powles, T.; Gallieni, M.; Bex, A. The adjuvant treatment of kidney cancer: A multidisciplinary outlook. Nat. Rev. Nephrol. 2019, 15, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zheng, X.; Du, Y.; Xing, Y.; Xu, K.; Cui, L. Matrix metalloproteinase-7 may serve as a novel biomarker for cervical cancer. OncoTargets Ther. 2018, 11, 4207–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Efferth, T.; Li, P.C.; Konkimalla, V.S.B.; Kaina, B. From traditional Chinese medicine to rational cancer therapy. Trends Mol. Med. 2007, 13, 353–361. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Kalscheuer, S.M.; Panyam, J. Exploiting nanotechnology to overcome tumor drug resistance: Challenges and opportunities. Adv. Drug Deliv. Rev. 2013, 65, 1731–1747. [Google Scholar] [CrossRef] [Green Version]
- Katz, O.B.; Shaked, Y. Host effects contributing to cancer therapy resistance. Drug Resist. Updat. 2015, 19, 33–42. [Google Scholar] [CrossRef]
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updat. 2017, 31, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.; Singh, A.; Nazir, S.U.; Tulsyan, S.; Khan, A.; Kumar, R.; Bashir, N.; Tanwar, P.; Mehrotra, R. Cancer drug resistance: A fleet to conquer. J. Cell. Biochem. 2019, 120, 14213–14225. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Ren, J.; Tang, J.; Zhang, D.; Li, B.; Li, Y. Estrogen-like activities of saikosaponin-d in vitro: A pilot study. Eur. J. Pharmacol. 2010, 626, 159–165. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Cancer Type | Cell Line | Dose/Conc. | Exposure (Hours) | Effects on Signaling Pathways | Reference |
|---|---|---|---|---|---|---|
| 1. | Lung cancer | A549 | 1–20 μM | 24 h | ♦ Fas/FasL apoptotic system and the p53 pathway, ◙ G1-phase ◘, proliferation restraint. | [48] |
| 2. | Lung cancer | A549, H1299 | 5–20 μM | 24 h | ◙ STAT3 pathway, ◘ G0/G1-phase arrest, ◙ proliferation, ◘ apoptosis. | [49] |
| 3. | Lung cancer | HCC827, H1975, PC-9, HCC827/GR | 5–40 μM | 24 h | ◙ STAT3 pathway, ◙ proliferation, ◘ apoptosis, Chemosensitization (gefitinib). | [50] |
| 4. | Lung cancer | A549 | 2 μM | 48 h | ◘ ROS accumulation, Enhancement of apoptosis, Chemosensitization (CDDP). | [51] |
| 5. | Lung cancer | A549 | 0.5, 2 μM | 12–48 h | ◙ Proliferation and migration by diminishing the JNK/pJNK negatively regulating p53, ◙ G1 and G2 cell cycle. | [52] |
| 6. | Lung cancer | HELF | 2.5, 5, 10 μg/mL | 96 h | ◙ Proliferation and TGF-β1 expression, ◙ epithelium-mesenchymal transition and alveolar epithelial cells. | [53] |
| 7. | Breast cancer | MCF-7, 4T-1, RAW 264.7 LO-2 | 0–60 μM | 24 or 48 h | ◘ Cell death, ◙ PI3K/Akt and MAPK/ERK pathways. | [54] |
| 8. | Breast cancer | HCC1937 | 13–100 μM | 2–24 h | ◙ Wnt/β-catenin pathway, ◙ proliferation, ◘ apoptosis. | [55] |
| 9. | Breast cancer | SK-BR-3, MCF -7, HBL-100 | 2.5, 5, and 10 mM | 48 h. | Bcl - 2, proto-oncogenetyrosine-protein kinase src are regulated, ◙ cell proliferation by estrogen receptors. | [56] |
| 10. | Breast cancer | MDA-MB-231 | 6–15 μM | 24 h | ♦ p38 pathway, ◙ viability, ◘ apoptosis. | [37] |
| 11. | Breast cancer | MCF-7 | 40 μg/mL | 72 h | ◙ P-gp in MCF-7/Pac, amplify the antiproliferative impact, MDR reversal in MCF-7 sublines that are resistant. | [57] |
| 12. | Breast cancer | MCF-7 | 10 μM | 4 h | ◙ SERCA, ♦ CaMKKβ-AMPK-mTOR signaling cascade, ER stress, and UPR, ◘ apoptosis and autophagy. | [38] |
| 13. | Breast cancer | MCF-7/ADR | 0.13–0.6 μM | 48 h | ◙ P-gp expression, MDR reversal without harmful consequences, Chemosensitization (doxorubicin). | [58] |
| 14. | Breast cancer | MCF-7/ADR, MCF-7 | 0.13–0.6 μM | 48 h | ▼MDR1/P-gp, Reversal of MDR without toxic effect, Chemosensitization (ADR). | [59] |
| 15. | Breast cancer | MDA-MB-231 | 6.25 μmol/L–12.50 μmol/L | 12, 24, 48 h | ◙ MDA-MB-231 cells in G2 phase, cell cycle arrest, ▼cyclin A1, cyclin A2, cyclin B1, and cyclin B2. | [41] |
| 16. | Liver cancer | SMMC-7721 | 3.2–19.2 μM | 24 h, 48 h, 72 h | ◙ p-STAT3/HIF-1α pathway and ◘ Suppression of COX-2 expression, ◙ proliferation. | [60] |
| 17. | Liver cancer | SMMC-7721, MHCC97L | 3 μg/mL | 2 h | ▲ Radiation, ◘ apoptosis by promoting autophagy via ◙ mTOR phosphorylation | [61] |
| 18. | Liver cancer | SMMC-7721, HepG2 | 3.2–19.2 μM | 24 h, 48 h, 72 h | ◙ p-STAT3/C/EBPβ pathway and COX-2 expression, ◙ proliferation, ◘apoptosis. | [47] |
| 19. | Liver cancer | HepG2 | 1.75, 3.75, 7.5, 15 μM | 24 h | NRP -1 knockdown dramatically changed the lipid transport, phospholipid metabolism, and enhanced anti-hepatoma action. | [62] |
| 20. | Liver cancer | HepG2, Hep3B | 1–10 μM | 12 h–48 h | ♦ p53 and Fas/FasL pathway, ◙ NF-κB pathway, ◘ G1- cell cycle arrest, ◙ proliferation, ◘ apoptosis. | [63] |
| 21. | Liver cancer | HepG2 | 2.5, 5 and 10 μM | 16 h | ◙ Hepatic FA biosynthesis, PPAR activation modifies the INSIGs/SREBP1c pathway. | [64] |
| 22. | Liver cancer | HepG2 | 10 μM | 24 h | ◙ NF-κB ♦, ◙ proliferation, angiogenesis, and invasion, ◘ apoptosis. | [34] |
| 23. | Liver cancer | Hepatocyte | 100, 200, and 400 ng/mL | 24 h | Enhance cell viability, ◙ apoptosis, ◙ mortality of hepatocytes. | [65] |
| 24. | Liver cancer | SMMC-7721 HepG2 | 1.28, 3.84 μM 3 μg/ml | 2 h | ♦ p53 pathway, ▲ G0/G1 arrest, ◘ G2/M-phase arrest under hypoxia, ◘ apoptosis, Radiosensitization. Bax▲, HIF-1α▼. | [40] |
| 25. | Liver cancer | Hep3B | 5 μM | 24 h | ◘ Cell apoptosis via both mechanisms reliant on and independent of caspase-3. | [66] |
| 26. | Liver cancer | SMMC-7721, MHCC97L | 3.84 μM | 24 h, 48 h, and 72 h | Suppression of mTOR pathway, ◙ proliferation, ◘ autophagy formation, radiosensitization. | [67] |
| 27. | Liver cancer | Hep3B | 2–15 μM | 48 h | ▲SENP5 expression and subsequent ◙ Gli1 SUMOylation, ◙ SHh pathway, ◙ viability, invasion and migration, ◘ apoptosis, chemosensitization (HSVtk/GCV). | [68] |
| 28. | Liver cancer | HepG2 | 5 μg/mL–20 μg/mL | 6 h | ◘ Apoptosis, ♦ caspases-3 and caspases-7. | [69] |
| 29. | Liver cancer | HSC-T6 LO2 | 1 μM 0.4 μM | 24 h | ▼ Expression of a smooth muscle actin, TGF-β1, ERK1/2, PDGFR, TGF-β1R, extracellular matrix regulated kinase 1, and connective tissue growth factor. | [70] |
| 30. | Liver cancer | SMMC-7721 | 5.0, 7.5, 10.0, 12.5, 15.0, and 17.5 mg/L | 24 h, 48 h, and 72 h | ◘ Autophagy through ▲ expression of BECN1, ◙ SMMC-7721 proliferation. | [71] |
| 31. | Liver cancer | Hep3B | - | - | Hep3B cells may be made to undergo apoptosis by using caspase-3-independent mechanisms. | [72] |
| 32. | Kidney cancer | Mouse renal tubular epithelial (mTE) cells | - | - | Prevents AKI via ◙ NLRP3 inflammasome by SIRT1, ROS ◙ | [73] |
| 33. | Kidney cancer | LLC-PK1 | 1 or 3 μg/mL | 24 h | ▲ Expression of anti-oxidant enzymes (SOD, CAT, GPx) and HSP72. | [74] |
| 34. | Kidney cancer | UCL93 and OX161 | 5.0 μM | 24 h | CaMKK-AMPK-mTOR signaling pathway to ◘ autophagy, ◙ SERCA to increase calcium levels. | [75] |
| 35. | Kidney cancer | NRK-52E | 45 and 60 μM | 24 h or 48 h | Attenuates oxidative injury via ▲ of SirT3, SOD activity▼ and SIRT3 expression ▲. | [76] |
| 36. | Kidney cancer | HK-2 | 20–150 μM | 0–48 h | ROS-mediated ♦ of MAPK and NF-κB signal pathways. | [77] |
| 37. | Kidney cancer | 769-P, 786-O | 10–20 μM | 48 h | ◙ EGFR/p38 pathway, ▲ p53. ◘ apoptosis, ◘ G0/G1-phase arrest, ◙ proliferation. | [78] |
| 38. | Cervical cancer | HeLa, Siha | 2 μM | 24 h, 36 h | ◘ Intracellular ROS accumulation, Enhancement of apoptosis, Chemosensitization (CDDP). | [51] |
| 39. | Cervical cancer | HeLa | 10 μM | 24 h | ◙ SERCA, ♦ CaMKK-AMPK-mTOR kinase signaling cascade, ER stress and UPR, ◘ apoptosis and autophagy. | [38] |
| 40. | Cervical cancer | HeLa | 10 μM | 0–24 h | ◙ NF-κB pathway, ◙ proliferation, angiogenesis and invasion. ◘ apoptosis, Chemosensitization (TNF-α). | [34] |
| 41. | Blood cancer | NB4, Kas-1, MV4-11, and U937 | 0.5 to 1 μM | 48 h | By concentrating on FTO/m6A and its ancillary pathways and ◙ AML leukemogenesis. | [79] |
| 42. | Blood cancer | HL60 | 12.8–19.2 μM | 48 h | ▲GR mRNA expression, ◘ G0/G1- phase arrest, ◙ proliferation. | [80] |
| 43. | Blood cancer | NB4 | 1.56, 3.12, and 6.25 μg/mL | 5 days | ▲Bactericidal activity, ◘ granulocyte differentiation via ▲PU.1, CEBPβ, and activating CBL-ERK1/2 pathway, ◙ proliferation. | [81] |
| 44. | Blood cancer | THP-1 | 1.8, 3.0, and 4.3 μM | 48 h | ◙ Selectin-mediated cell adhesion. | [82] |
| 45. | Brain cancer | U87 | 1–8 μM | 48 h | ▼ PI3K/Akt and ERK pathway, ♦ JNK, ◙ proliferation, Enhancement of apoptosis. | [83] |
| 46. | Brain cancer | Primary microglia cells | 0, 0.1, 0.25, 0.5, 1, 2, 4 μM | 24 h | ◘ Acute inflammatory depressive-like behaviors and microglia ♦, ◙ downstream TLR4/ NF-κB pathway. | [24] |
| 47. | Brain cancer | C6 | 2.8–128 μM | 4 days | ◘ Differentiation, ◙ growth. | [84] |
| 48. | Brain cancer | Shh Light II and DAOY | 3 μM | 36 h | ◙ Cell proliferation, ▼ mRNA in Gli1 and Ptch1, GLI-luciferase activity and Hh signaling. | [85] |
| 49. | Brain cancer | C6 rat glioma cells | 1–20 μM [PGE2] 10–100 μM [Ca2+] | 24 h | ◙ PGE2 production, ◙ cyclooxygenase activity, and an elevation of [Ca2+]. | [86] |
| 50. | Brain cancer | PC12 | 0.125–2 μg/mL | 24 h | Controls nuclear and mitochondrial GR translocation, partially reversing mitochondrial dysfunction, ◙ mitochondrial apoptotic pathway, ♦ GR-dependent survival pathway. | [87] |
| 51. | Brain cancer | PC12 | 200, 300, and 400 μg/mL | 6–96 h | ▼PC12 cells’ apoptosis by reducing ROS and ◙ oxidative damage caused by MAPK. | [29] |
| 52. | Ovarian cancer | SKOV3 | 2 μM | 48 h | ◘ Intracellular ROS accumulation, Enhancement of apoptosis, Chemosensitization (CDDP). | [51] |
| 53. | Ovarian cancer | A2780s, A2780cp, Hey, SKOV3 | 1, 2 μM | 24 h | ▲ Ca2+ concentration, ◘ MMP loss, ♦ CaMKI, ◙ PPM1D, Promotion of mitochondrial fission, ◘ G2/M arrest. Chemosensitization (CDDP). | [88] |
| 54. | Prostate cancer | DU145 | 2.5–50 μM | 24 h | ▲ p53, ◙ proliferation, ◘ G0/G1-phase arrest, ◘ of apoptosis. | [89] |
| 55. | Prostate cancer | DU145, CWR22Rv1 | 5, 10 μM | 24 h, 48 h, 72 h | ◙ GSK3β/β-catenin pathway in CWR22Rv1, Suppression of proliferation, metastasis and invasion. | [90] |
| 56. | Osteosarcoma | 143B, MG-63 | 80 μM | 24 h, 48 h, 72 h | ♦ p53 pathway, ◘ apoptosis, ◘ G0/G1-phase arrest, ◙ proliferation. | [91] |
| 57. | Osteosarcoma | U2 | 5–20 μM | 24 h, 36 h, 48 h | ◙ Akt and ERK pathway, ◙ proliferation, invasion, and migration, ◘ apoptosis. | [92] |
| 58. | Colon cancer | SW480 and SW620 | 50 μg/mL | 24 h | Promote apoptosis, ◙ PI3K/Akt/mTOR pathway and proliferation. | [93] |
| 59. | Colon cancer | HT-29 | 10 μg/mL | 24 h | Apoptosis of HT29 ▲, TRAIL, TRAIL-R and caspase10 and/or caspase8 ▼. | [94] |
| 60. | Lymphoid tissue | Mouse T cells | 5–15 μM | 48 h | ◙ T cell proliferation, ♦ NF-κB, NF-AT, and AP-1 signal pathways, ◙ cytokine secretion, IL-2 receptor expression. | [95] |
| 61. | Lymphoid tissue | Rat basophilic leukemia-2H3 cells | 50 μg/mL | 1 h | ◙ Intracellular calcium mobilization, ROS, cell degranulation, and tyrosine phosphorylation, ◙ gene ♦ of Cdc42 and c-Fos. | [96] |
| 62. | Thyroid carcinoma | ARO, 8305C, SW1736 | 5–20 μM | 12 h, 24 h, 48 h | ♦ p53 pathway, ◙ proliferation, ◘ G1-phase arrest, ◘ apoptosis. | [97] |
| 63. | Melanoma | A375.S2 | 5–20 μM | 30 min | ♦ JNK, p38, and p53, ◙ proliferation, ◘ apoptosis. | [98] |
| 64. | Pancreatic cancer | BxPC3 | 1–8 μM | 48 h and 72 h | ♦ MKK4-JNK pathway, ◙ proliferation, ◘ apoptosis. | [99] |
| 65. | Gastric cancer | SGC-7901, MGC-803, and HGC-27 | 2.5 μg/mL | 72 h | ◙ IKK β/NF-κB pathway, ◘ both cell autophagy and apoptosis. | [100] |
| S. No. | Cancer Type | Model | Dose/Conc. | Exposure (Days) | Route of Administration | Effects on Signaling Pathways | Reference |
|---|---|---|---|---|---|---|---|
| 1. | Lung cancer | HCC827/GR cells xenograft tumor in nude mice | 5, 10 mg/kg | 14 days | Intraperitoneal (IP) | ◙ Growth, ◘ apoptosis, Chemosensitization (gefitinib) | [50] |
| 2. | Lung cancer | BLM (5 mg/kg)-induced PF mice | 2 mg/kg | 28 days | IP | Alleviated pulmonary alveolitis, pulmonary fibrosis and cell apoptosis. ◙ Caspase-3, FN, Wnt and β-catenin, E-cad upregulated. | [53] |
| 3. | Lung cancer | VILI rats | IP | ▼ MIP-2, IL-6, and TNF-α and ▲ TGF-β1 and IL-10. | [101] | ||
| 4. | Breast cancer | BALB/c mice (female, 4–6 weeks old) | 1 or 5 mg/kg | 3 days | Tail vein | ◙ p-Akt and p-ERK. | [54] |
| 5. | Breast cancer | MCF-7/ADR cells xenograft tumor in nude mice | 5 mg/kg | 20 days | IP | ◙ Growth, ◙ P-gp expression, Reversal of MDR without toxic effect. | [58] |
| 6. | Liver cancer | HSVtk/Hep3B cells xenograft tumor in nude mice | 10 mg/kg | 33 days | IP | ◙ Growth, ◘ apoptosis, Chemosensitization (HSVtk/GCV) | [68] |
| 7. | Liver cancer | Eight-week-old male C57BL/6J mice. | 5, 10, and 20 mg/kg | 4 weeks | Intragastric (IG) | ◙ FA synthesis by retaining SREBP1c, ♦ INSIG1, INSIG2, and PPARα. ◘ FA catabolism in WAT. | [64] |
| 8. | Liver cancer | BALB/c nude mice bearing SMMC-7721 xenograft tumor, | 0.75 mg/kg | Thrice a week for 2 weeks | IP | ◙ HIF-1α | [102] |
| 9. | Liver cancer | Fish–hybrid grouper | 100, 200, 400, and 800 mg/kg | 56 days | IP | ▼ IL-6, TNF-α, and IL-1β levels in liver tissue, and markedly immune inflammatory response and ◙ apoptosis. | [65] |
| 10. | Liver cancer | DEN-induced Sprague Dawley rat HCC model | 2 mg/kg | 17 weeks | IP | ◙ C/EBPβ and COX-2 | [103] |
| 11. | Liver cancer | C57/BL6rats | 2 mg/kg 0.3 and 0.6 μg/mL | 5 days | IP | ◙ NF-κB and STAT3-mediated inflammatory signal pathway. | [44] |
| 12. | Liver cancer | Hepatic fibrosis rats | 1.0, 1.5, and 2.0 mg/kg | 6 weeks | IP | ◙ Liver TNF- α, IL-6, and NF-κB p65 expression and ▲ I-κBα activity. | [104] |
| 13. | Liver cancer | SD rats | 0.03% SSD | 16 weeks | IG | ▼Syndecan-2, MMP-2, MMP-13, and TIMP-2 tissue. | [33] |
| 14. | Liver cancer | SD rats | 0.75, 1.50 mg/kg | ▼Serum corticosterone levels, BDNF, neurons generations, GR expression, and nuclear translocation▲. | [105] | ||
| 15. | Liver cancer | SD male rats | 1.0 mg/kg | Once a day for 18 days | IP | ◙ Angiogenesis of DEN-induced hepatocarcinogenesis, ◙ Ang-2, and VEGF. | [106] |
| 16. | Kidney cancer | Male C57BL/6J mice | 10 mg/kg | 3 days | IP | ▼ Kidney injury and inflammation, ◘ SIRT1, ◙ IL-1B, NLRP3, SIRT1, and ROS. | [73] |
| 17. | Kidney cancer | Wistar rats–Anti-Thy1 mAb 1–22–3-induced rat model of glomerulonephritis | 0.6 or 1.8 mg/kg | 31 days | IP | ▼ TGF-β1 and type I collagen. | [107] |
| 18. | Kidney cancer | LPS-induced mice | 5, 20 mg/kg | 1 week | IG | ◙ FOSL1, TCF7, ◙ MMP9 expression and ▼ renal inflammation and ◘ cell apoptosis. | [108] |
| 19. | Blood cancer | C57BL6/J mice | 6, 12 mg/kg | 6 days | IP | ▲ Bactericidal activity, ◘ granulocytic differentiation by ♦ CBL-ERK1/2 pathway. | [81] |
| 20. | Anti-Tumor activity | A549 cells-bearing nude mice | 1.0 mg/kg | IG | ◘ Apoptosis, ◙ COX-2. | [109] | |
| 21. | Brain cancer | MB allo-graft mice | 10 mg/kg | 18 days | IP | ◙ Tumor growth, ▼Gli1, mRNA, ◙ Hh signaling pathway by targeting SMO. | [85] |
| 22. | Brain cancer | Male ICR mice (18–22 g; 6–8 weeks old) | 1 mg/kg | 7 days | IG | ▲LPS-induced inflammation, ◙ LPS-induced microglia activation and neuroinflammation. ▼HMGB1/TLR4/NF-κB. | [24] |
| 23. | Brain cancer | 3xTg mice (age, 9 months; weight, 30–35 g) | 10 mg/kg | 28 days | Oral | ▼Cell apoptosis and inflammation, ◙ NF-κB activation. ◙ activation of microglia and astrocytes. | [110] |
| 24. | Thyroid cancer | ARO cells xenograft tumor in nude mice | 5–20 mg/kg | 4 weeks | Oral | ◙ Tumor growth. | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manoharan, S.; Deivendran, B.; Perumal, E. Chemotherapeutic Potential of Saikosaponin D: Experimental Evidence. J. Xenobiot. 2022, 12, 378-405. https://doi.org/10.3390/jox12040027
Manoharan S, Deivendran B, Perumal E. Chemotherapeutic Potential of Saikosaponin D: Experimental Evidence. Journal of Xenobiotics. 2022; 12(4):378-405. https://doi.org/10.3390/jox12040027
Chicago/Turabian StyleManoharan, Suryaa, Bhuvaneshwari Deivendran, and Ekambaram Perumal. 2022. "Chemotherapeutic Potential of Saikosaponin D: Experimental Evidence" Journal of Xenobiotics 12, no. 4: 378-405. https://doi.org/10.3390/jox12040027