

Gene Therapy for Inherited Hearing Loss: Updates and Remaining Challenges
Abstract
:1. Introduction
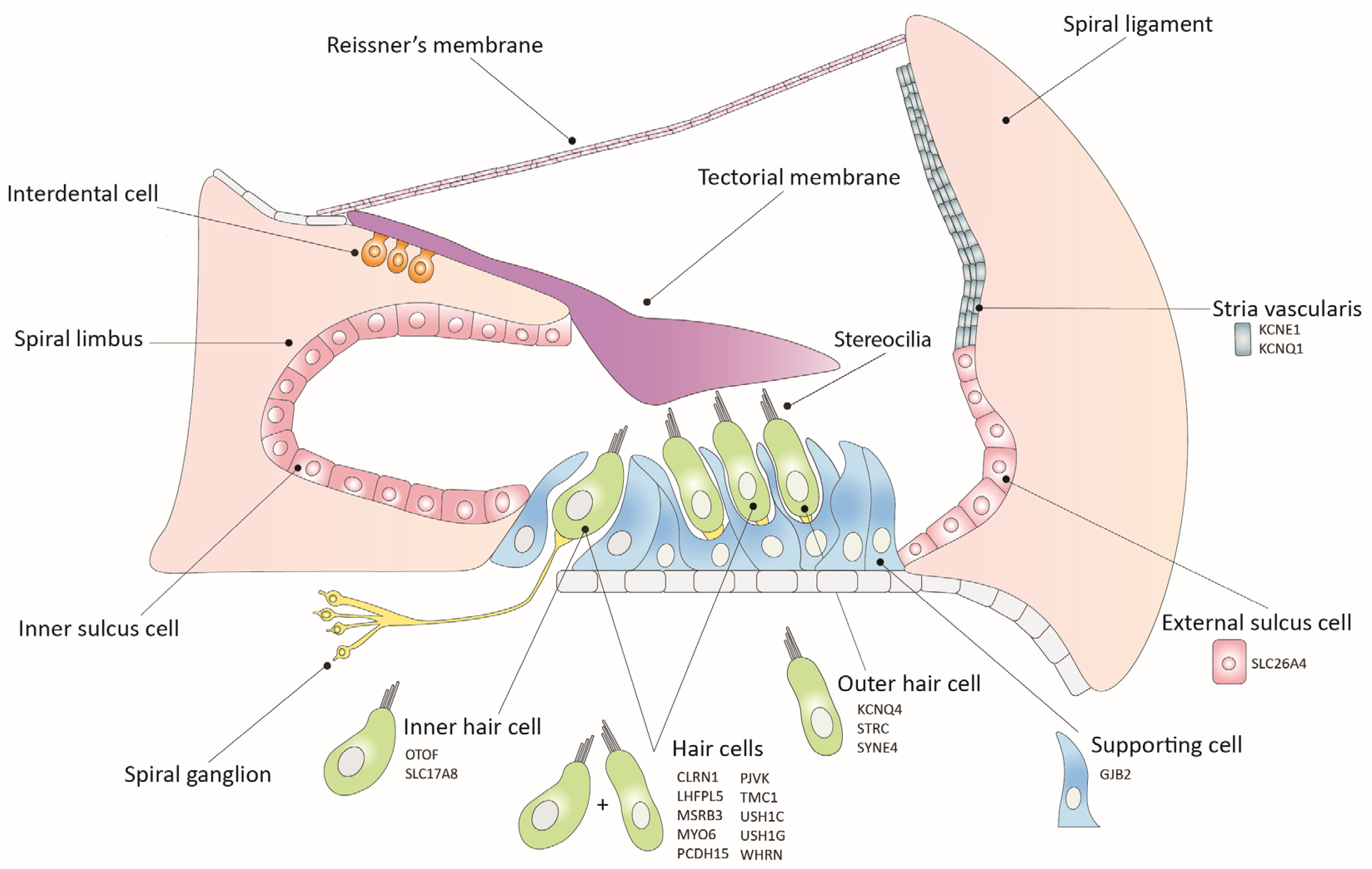
2. Inner Ear Gene Therapy Strategies
2.1. Gene Replacement
2.2. Gene Suppression

{kind=link}
{kind=link}
| Gene (Deafness Form) | Animal Model | Hearing Impairment | Strategy | Injection Age | Injection Route | Vector | Reference |
|---|---|---|---|---|---|---|---|
| GJB2 (DFNA3A) | Gjb2 p.R75W | Severe to profound | RNAi | P42–P45 | RWM | siRNAs | [49] |
| TMC1 (DFNA36) | Tmc1-Bth | Progressive | miRNA | P0–P2 | RWM | AAV9 | [50] |
| Tmc1-Bth | Progressive | miRNA | P15–P16; P56–P60; P84–P90 | RWM, PSCC | AAV9 | [51] | |
| Tmc1-Bth | Progressive | RNA editing (CasRx) | P1–P2 | RWM | AAV9-PHP.eB | [52] | |
| USH1C (USH1C) | Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | P1; P3; P5; P7 | Intraperitoneal | ASO-29 | [58] |
| Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | P1; P5; P10; P20 | RWM | ASO-29 | [56] | |
| Ush1c c.216G>A | Severe (balance defect) | Antisense oligonucleotide | E12.5 | EUGO | ASO-29 | [59] |
2.3. Gene Editing
| Gene (Deafness Form) | Animal Model | Hearing Impairment | Strategy | Injection Age | Injection Route | Vector | Reference |
|---|---|---|---|---|---|---|---|
| KCNQ4 (DFNA2A) | Kcnq4 c.827G>C | Progressive | Disruption | P0–P1 | PSCC, RWM, utricle, scala media | Dual vector: Anc80L65 | [69] |
| Kcnq4 c.683G>A | Progressive | Disruption | P1–P2 | Scala media | AAV9-PHP.eB | [65] | |
| MYO6 (DFNA22) | Myo6 p.C442Y | Progressive | Disruption | P0–P2 | Scala media | AAV9-PHP.eB- | [70] |
| Myo6 p.C442Y | Progressive | RNA base editing | P0–P2 | Scala media | AAV9-PHP.eB (RNA ABE) | [68] | |
| PCDH15 (DFNB23/USH1F) | Pcdh15av−3J | Profound (balance defect) | Frame restoration | P0–P2 | Scala media | AAV9 | [64] |
| TMC1 (DFNA36) | Tmc1-Bth | Progressive | Disruption | P1 | Utricle | Dual vector: AAV9-PHP.B | [40] |
| Tmc1-Bth | Progressive | Disruption | P1–P2 | Inner ear | Dual vector: Anc80 L65 | [71] | |
| TMC1 (DFNB7/11) | Tmc1-Baringo | Profound | Base editing | P1 | Inner ear | Dual vector: Anc80 L65 (CBE) | [67] |
3. Delivery Vectors
3.1. Adeno-Associated Virus
3.2. Lentivirus
3.3. Adenovirus
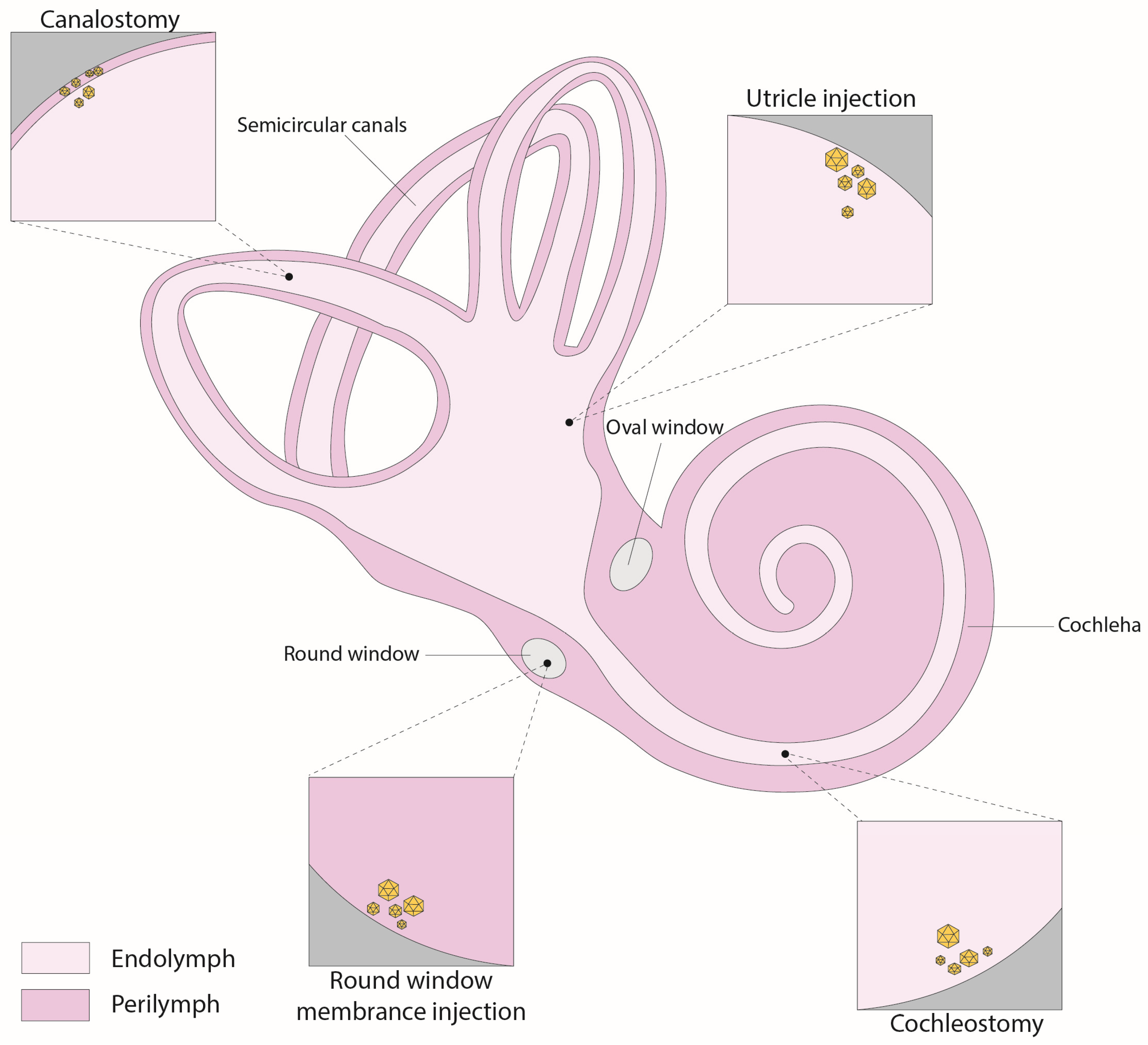
4. Inner Ear Delivery Approaches
4.1. Round-Window Injection
4.2. Canalostomy
4.3. Cochleostomy
4.4. Utricle
5. Challenges and Limitations
5.1. The Mouse as a Model for Human Deafness
5.2. Genetic Heterogenicity
5.3. Applications in Mature Mice
5.4. Implications of the Immune Response
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheffield, A.M.; Smith, R.J.H. The epidemiology of deafness. Cold Spring Harb. Perspect. Med. 2019, 9, a033258. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, B.O.; Davis, A.C.; Hoffman, H.J. Hearing loss: Rising prevalence and impact. Bull. World Health Organ. 2019, 97, 646. [Google Scholar] [CrossRef]
- Carpena, N.T.; Lee, M.Y. Genetic hearing loss and gene therapy. Genom. Inform. 2018, 16, e20. [Google Scholar] [CrossRef] [PubMed]
- Alford, R.L.; Arnos, K.S.; Fox, M.; Lin, J.W.; Palmer, C.G.; Pandya, A.; Rehm, H.L.; Robin, N.H.; Scott, D.A.; Yoshinaga-Itano, C.; et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 2014, 16, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, D.; He, Y.; Shu, Y. Advances in gene therapy hold promise for treating hereditary hearing loss. Mol. Ther. 2023, 31, 934–950. [Google Scholar] [CrossRef] [PubMed]
- Geleoc, G.S.; Holt, J.R. Sound strategies for hearing restoration. Science 2014, 344, 1241062. [Google Scholar] [CrossRef]
- Delmaghani, S.; El-Amraoui, A. Inner ear gene therapies take off: Current promises and future challenges. J. Clin. Med. 2020, 9, 2309. [Google Scholar] [CrossRef]
- Petit, C.; Bonnet, C.; Safieddine, S. Deafness: From genetic architecture to gene therapy. Nat. Rev. Genet. 2023, 24, 665–686. [Google Scholar] [CrossRef]
- Amariutei, A.E.; Jeng, J.Y.; Safieddine, S.; Marcotti, W. Recent advances and future challenges in gene therapy for hearing loss. R. Soc. Open Sci. 2023, 10, 230644. [Google Scholar] [CrossRef]
- Klimara, M.J.; Smith, R.J.H. Advances in cochlear gene therapies. Curr. Opin. Pediatr. 2023, 35, 631–640. [Google Scholar] [CrossRef]
- Dror, A.A.; Avraham, K.B. Hearing impairment: A panoply of genes and functions. Neuron 2010, 68, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Taiber, S.; Gwilliam, K.; Hertzano, R.; Avraham, K.B. The genomics of auditory function and disease. Annu. Rev. Genom. Hum. Genet. 2022, 23, 275–299. [Google Scholar] [CrossRef]
- Muller, U.; Barr-Gillespie, P.G. New treatment options for hearing loss. Nat. Rev. Drug Discov. 2015, 14, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Shubina-Oleinik, O.; Holt, J.R. Emerging gene therapies for genetic hearing loss. J. Assoc. Res. Otolaryngol. 2017, 18, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Taiber, S.; Avraham, K.B. Genetic therapies for hearing loss: Accomplishments and remaining challenges. Neurosci. Lett. 2019, 713, 134527. [Google Scholar] [CrossRef]
- Askew, C.; Chien, W.W. Adeno-associated virus gene replacement for recessive inner ear dysfunction: Progress and challenges. Hear. Res. 2020, 394, 107947. [Google Scholar] [CrossRef]
- Lahlou, G.; Calvet, C.; Giorgi, M.; Lecomte, M.J.; Safieddine, S. Towards the clinical application of gene therapy for genetic inner ear diseases. J. Clin. Med. 2023, 12, 1046. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Hathaway, D.M.; Klein, A.J.; Peters, C.W.; Li, Y.; Tamvakologos, P.I.; Nammour, J.; Maguire, C.A.; Corey, D.P. AAV-S: A versatile capsid variant for transduction of mouse and primate inner ear. Mol. Ther. Methods Clin. Dev. 2021, 21, 382–398. [Google Scholar] [CrossRef]
- Geng, R.; Omar, A.; Gopal, S.R.; Chen, D.H.; Stepanyan, R.; Basch, M.L.; Dinculescu, A.; Furness, D.N.; Saperstein, D.; Hauswirth, W.; et al. Modeling and preventing progressive hearing loss in usher syndrome iii. Sci. Rep. 2017, 7, 13480. [Google Scholar] [CrossRef]
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, P.F.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of usher syndrome. J. Clin. Investig. 2018, 128, 3382–3401. [Google Scholar] [CrossRef]
- Gyorgy, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of Usher Syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ma, X.; Skidmore, J.M.; Cimerman, J.; Prieskorn, D.M.; Beyer, L.A.; Swiderski, D.L.; Dolan, D.F.; Martin, D.M.; Raphael, Y. GJB2 gene therapy and conditional deletion reveal developmental stage-dependent effects on inner ear structure and function. Mol. Ther. Methods Clin. Dev. 2021, 23, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Kamiya, K.; Gotoh, S.; Sugitani, Y.; Suzuki, M.; Noda, T.; Minowa, O.; Ikeda, K. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum. Mol. Genet. 2015, 24, 3651–3661. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.; Li, Y.; Zhang, W.; Wang, J.; Cai, C.; Lin, X. Gene therapy via canalostomy approach preserves auditory and vestibular functions in a mouse model of Jervell and Lange-Nielsen Syndrome Type 2. Nat. Commun. 2021, 12, 697. [Google Scholar] [CrossRef]
- Chang, Q.; Wang, J.; Li, Q.; Kim, Y.; Zhou, B.; Wang, Y.; Li, H.; Lin, X. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 2015, 7, 1077–1086. [Google Scholar] [CrossRef]
- Gyorgy, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of hearing by gene delivery to inner-ear hair cells using exosome-associated AAV. Mol. Ther. 2017, 25, 379–391. [Google Scholar] [CrossRef]
- Kim, M.A.; Cho, H.J.; Bae, S.H.; Lee, B.; Oh, S.K.; Kwon, T.J.; Ryoo, Z.Y.; Kim, H.Y.; Cho, J.H.; Kim, U.K.; et al. Methionine sulfoxide reductase b3-targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid. Redox Signal. 2016, 24, 590–602. [Google Scholar] [CrossRef]
- Tang, H.; Wang, H.; Wang, S.; Hu, S.W.; Lv, J.; Xun, M.; Gao, K.; Wang, F.; Chen, Y.; Wang, D.; et al. Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum. Genet. 2023, 142, 289–304. [Google Scholar] [CrossRef]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; Boutet de Monvel, J.; Hardelin, J.P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kugler, S.; Reisinger, E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf Otoferlin knock-out mice. EMBO Mol. Med. 2019, 11, e9396. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hathaway, D.M.; Klein, A.J.; Pan, B.; Strelkova, O.; De-la-Torre, P.; Wu, X.; Peters, C.W.; Mulhall, E.M.; Booth, K.T.; et al. Mini-Pcdh15 gene therapy rescues hearing in a mouse model of Usher Syndrome type 1F. Nat. Commun. 2023, 14, 2400. [Google Scholar] [CrossRef]
- Lu, Y.C.; Tsai, Y.H.; Chan, Y.H.; Hu, C.J.; Huang, C.Y.; Xiao, R.; Hsu, C.J.; Vandenberghe, L.H.; Wu, C.C.; Cheng, Y.F. Gene therapy with a synthetic adeno-associated viral vector improves audiovestibular phenotypes in Pjvk-mutant mice. JCI Insight 2022, 7, e152941. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, B.K.; Miyakoshi, L.M.; Cederroth, C.R.; Tserga, E.; Versteegh, C.; Bork, P.A.R.; Hauglund, N.L.; Gomolka, R.S.; Mori, Y.; Edvall, N.K.; et al. Delivery of gene therapy through a cerebrospinal fluid conduit to rescue hearing in adult mice. Sci. Transl. Med. 2023, 15, eabq3916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, H.; Liu, H.; Cai, R.; Wu, H. Gene therapy restores auditory functions in an adult Vglut3 knockout mouse model. Hum. Gene Ther. 2022, 33, 729–739. [Google Scholar] [CrossRef]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of hearing in the Vglut3 knockout mouse using virally mediated gene therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef]
- Kim, M.A.; Kim, S.H.; Ryu, N.; Ma, J.H.; Kim, Y.R.; Jung, J.; Hsu, C.J.; Choi, J.Y.; Lee, K.Y.; Wangemann, P.; et al. Gene therapy for hereditary hearing loss by SLC26A4 mutations in mice reveals distinct functional roles of pendrin in normal hearing. Theranostics 2019, 9, 7184–7199. [Google Scholar] [CrossRef]
- Shubina-Oleinik, O.; Nist-Lund, C.; French, C.; Rockowitz, S.; Shearer, A.E.; Holt, J.R. Dual-vector gene therapy restores cochlear amplification and auditory sensitivity in a mouse model of DFNB16 hearing loss. Sci. Adv. 2021, 7, eabi7629. [Google Scholar] [CrossRef]
- Taiber, S.; Cohen, R.; Yizhar-Barnea, O.; Sprinzak, D.; Holt, J.R.; Avraham, K.B. Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol. Med. 2021, 13, e13259. [Google Scholar] [CrossRef]
- Wu, J.; Solanes, P.; Nist-Lund, C.; Spataro, S.; Shubina-Oleinik, O.; Marcovich, I.; Goldberg, H.; Schneider, B.L.; Holt, J.R. Single and dual vector gene therapy with AAV9-PHP.B rescues hearing in tmc1 mutant mice. Mol. Ther. 2021, 29, 973–988. [Google Scholar] [CrossRef]
- Marcovich, I.; Baer, N.K.; Shubina-Oleinik, O.; Eclov, R.; Beard, C.W.; Holt, J.R. Optimized AAV vectors for TMC1 gene therapy in a humanized mouse model of DFNB7/11. Biomolecules 2022, 12, 914. [Google Scholar] [CrossRef] [PubMed]
- Nist-Lund, C.A.; Pan, B.; Patterson, A.; Asai, Y.; Chen, T.; Zhou, W.; Zhu, H.; Romero, S.; Resnik, J.; Polley, D.B.; et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; Boutet de Monvel, J.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1g. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [PubMed]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene therapy restores balance and auditory functions in a mouse model of Usher syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef]
- Maeda, Y.; Fukushima, K.; Nishizaki, K.; Smith, R.J. In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum. Mol. Genet. 2005, 14, 1641–1650. [Google Scholar] [CrossRef]
- Shibata, S.B.; Ranum, P.T.; Moteki, H.; Pan, B.; Goodwin, A.T.; Goodman, S.S.; Abbas, P.J.; Holt, J.R.; Smith, R.J.H. RNA interference prevents autosomal-dominant hearing loss. Am. J. Hum. Genet. 2016, 98, 1101–1113. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Moteki, H.; Smith, R.J.H. Targeted allele suppression prevents progressive hearing loss in the mature murine model of human TMC1 deafness. Mol. Ther. 2019, 27, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, G.; Cui, C.; Wang, F.; Wang, X.; Xu, Z.; Guo, H.; Chen, Y.; Tang, H.; Wang, D.; et al. Preventing autosomal-dominant hearing loss in bth mice with CRISPR/CasRx-based RNA editing. Signal Transduct. Target. Ther. 2022, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, S.; Depreux, F.F.; Jodelka, F.M.; Lentz, J.J.; Rigo, F.; Jones, T.A.; Hastings, M.L. Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum. Mol. Genet. 2017, 26, 3482–3494. [Google Scholar] [CrossRef]
- Robillard, K.N.; de Vrieze, E.; van Wijk, E.; Lentz, J.J. Altering gene expression using antisense oligonucleotide therapy for hearing loss. Hear. Res. 2022, 426, 108523. [Google Scholar] [CrossRef]
- Lentz, J.J.; Pan, B.; Ponnath, A.; Tran, C.M.; Nist-Lund, C.; Galvin, A.; Goldberg, H.; Robillard, K.N.; Jodelka, F.M.; Farris, H.E.; et al. Direct delivery of antisense oligonucleotides to the middle and inner ear improves hearing and balance in Usher mice. Mol. Ther. 2020, 28, 2662–2676. [Google Scholar] [CrossRef]
- Halloy, F.; Biscans, A.; Bujold, K.E.; Debacker, A.; Hill, A.C.; Lacroix, A.; Luige, O.; Stromberg, R.; Sundstrom, L.; Vogel, J.; et al. Innovative developments and emerging technologies in RNA therapeutics. RNA Biol. 2022, 19, 313–332. [Google Scholar] [CrossRef]
- Ponnath, A.; Depreux, F.F.; Jodelka, F.M.; Rigo, F.; Farris, H.E.; Hastings, M.L.; Lentz, J.J. Rescue of outer hair cells with antisense oligonucleotides in Usher mice is dependent on age of treatment. J. Assoc. Res. Otolaryngol. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Wang, L.; Kempton, J.B.; Jiang, H.; Jodelka, F.M.; Brigande, A.M.; Dumont, R.A.; Rigo, F.; Lentz, J.J.; Hastings, M.L.; Brigande, J.V. Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res. 2020, 48, 5065–5080. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Niggemann, P.; Gyorgy, B.; Chen, Z.Y. Genome and base editing for genetic hearing loss. Hear. Res. 2020, 394, 107958. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, E.; Bosio, A. The promise and the hope of gene therapy. Front. Genome Ed. 2021, 3, 618346. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR gene therapy: Applications, limitations, and implications for the future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zou, L.; Li, K.; Hou, H.; Hu, Q.; Liu, S.; Li, J.; Song, C.; Chen, J.; Wang, S.; et al. Template-independent genome editing in the Pcdh15av-3j mouse, a model of human DFNB23 nonsyndromic deafness. Cell Rep. 2022, 40, 111061. [Google Scholar] [CrossRef]
- Cui, C.; Wang, D.; Huang, B.; Wang, F.; Chen, Y.; Lv, J.; Zhang, L.; Han, L.; Liu, D.; Chen, Z.Y.; et al. Precise detection of CRISPR-Cas9 editing in hair cells in the treatment of autosomal dominant hearing loss. Mol. Ther. Nucleic Acids 2022, 29, 400–412. [Google Scholar] [CrossRef]
- Abdelnour, S.A.; Xie, L.; Hassanin, A.A.; Zuo, E.; Lu, Y. The potential of CRISPR/Cas9 gene editing as a treatment strategy for inherited diseases. Front. Cell Dev. Biol. 2021, 9, 699597. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.H.; Shubina-Oleinik, O.; Levy, J.M.; Pan, B.; Newby, G.A.; Wornow, M.; Burt, R.; Chen, J.C.; Holt, J.R.; Liu, D.R. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 2020, 12, eaay9101. [Google Scholar] [CrossRef]
- Xiao, Q.; Xu, Z.; Xue, Y.; Xu, C.; Han, L.; Liu, Y.; Wang, F.; Zhang, R.; Han, S.; Wang, X.; et al. Rescue of autosomal dominant hearing loss by in vivo delivery of mini dCas13X-derived RNA base editor. Sci. Transl. Med. 2022, 14, eabn0449. [Google Scholar] [CrossRef]
- Noh, B.; Rim, J.H.; Gopalappa, R.; Lin, H.; Kim, K.M.; Kang, M.J.; Gee, H.Y.; Choi, J.Y.; Kim, H.H.; Jung, J. In vivo outer hair cell gene editing ameliorates progressive hearing loss in dominant-negative Kcnq4 murine model. Theranostics 2022, 12, 2465–2482. [Google Scholar] [CrossRef]
- Xue, Y.; Hu, X.; Wang, D.; Li, D.; Li, Y.; Wang, F.; Huang, M.; Gu, X.; Xu, Z.; Zhou, J.; et al. Gene editing in a Myo6 semi-dominant mouse model rescues auditory function. Mol. Ther. 2022, 30, 105–118. [Google Scholar] [CrossRef]
- Gyorgy, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Zolotukhin, S. E pluribus unum: 50 years of research, millions of viruses, and one goal--tailored acceleration of aav evolution. Mol. Ther. 2015, 23, 1819–1831. [Google Scholar] [CrossRef]
- Keeler, A.M.; Flotte, T.R. Recombinant adeno-associated virus gene therapy in light of Luxturna (and Zolgensma and Glybera): Where are we, and how did we get here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef] [PubMed]
- Fakhiri, J.; Landegger, L.D.; Grimm, D. Breaking the sound barrier: Towards next-generation AAV vectors for gene therapy of hearing disorders. Hear. Res. 2022, 413, 108092. [Google Scholar] [CrossRef] [PubMed]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., 3rd; Strohl, W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Chien, W.W.; Monzack, E.L.; McDougald, D.S.; Cunningham, L.L. Gene therapy for sensorineural hearing loss. Ear Hear. 2015, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, W.; Zhang, Y.; Zidon, T.; Ritchie, T.; Engelhardt, J.F. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol. 1999, 73, 9468–9477. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Flotte, T.R. Gene therapy progress and prospects: Recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004, 11, 805–810. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Marrone, L.; Marchi, P.M.; Azzouz, M. Circumventing the packaging limit of AAV-mediated gene replacement therapy for neurological disorders. Expert. Opin. Biol. Ther. 2022, 22, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; Fan, P.D.; Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996, 7, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Nakai, H.; Xiao, W. Characterization of genome integrity for oversized recombinant AAV vector. Mol. Ther. 2010, 18, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Omichi, R.; Yoshimura, H.; Shibata, S.B.; Vandenberghe, L.H.; Smith, R.J.H. Hair cell transduction efficiency of single- and dual-AAV serotypes in adult murine cochleae. Mol. Ther. Methods Clin. Dev. 2020, 17, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.R.; Guo, J.Y.; He, L.; Liu, S.; Xu, J.Y.; Yang, Z.J.; Su, W.; Liu, K.; Gong, S.S.; Wang, G.P. Co-transduction of dual-adeno-associated virus vectors in the neonatal and adult mouse utricles. Front. Mol. Neurosci. 2022, 15, 1020803. [Google Scholar] [CrossRef] [PubMed]
- Akil, O. Dual and triple AAV delivery of large therapeutic gene sequences into the inner ear. Hear. Res. 2020, 394, 107912. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Wanisch, K.; Yanez-Munoz, R.J. Integration-deficient lentiviral vectors: A slow coming of age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Chang, Q.; Ahmad, S.; Zhou, B.; Kim, Y.; Li, H.; Lin, X. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J. Gene Med. 2013, 15, 123–133. [Google Scholar] [CrossRef]
- Han, J.J.; Mhatre, A.N.; Wareing, M.; Pettis, R.; Gao, W.Q.; Zufferey, R.N.; Trono, D.; Lalwani, A.K. Transgene expression in the guinea pig cochlea mediated by a lentivirus-derived gene transfer vector. Hum. Gene Ther. 1999, 10, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Tao, Y.; Li, W.; Shen, J.; Wang, Z.; Chen, Z.Y. Adenovirus vectors target several cell subtypes of mammalian inner ear in vivo. Neural Plast. 2016, 2016, 9409846. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gao, G. State-of-the-art human gene therapy: Part I. Gene delivery technologies. Discov. Med. 2014, 18, 67–77. [Google Scholar] [PubMed]
- Lasaro, M.O.; Ertl, H.C. New insights on adenovirus as vaccine vectors. Mol. Ther. 2009, 17, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes. Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Plontke, S.K.; Hartsock, J.J.; Gill, R.M.; Salt, A.N. Intracochlear drug injections through the round window membrane: Measures to improve drug retention. Audiol. Neurootol. 2016, 21, 72–79. [Google Scholar] [CrossRef]
- Xia, L.; Yin, S.; Wang, J. Inner ear gene transfection in neonatal mice using adeno-associated viral vector: A comparison of two approaches. PLoS ONE 2012, 7, e43218. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced viral-mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci. Rep. 2018, 8, 2980. [Google Scholar] [CrossRef]
- Kawamoto, K.; Oh, S.H.; Kanzaki, S.; Brown, N.; Raphael, Y. The functional and structural outcome of inner ear gene transfer via the vestibular and cochlear fluids in mice. Mol. Ther. 2001, 4, 575–585. [Google Scholar] [CrossRef]
- Talaei, S.; Schnee, M.E.; Aaron, K.A.; Ricci, A.J. Dye tracking following posterior semicircular canal or round window membrane injections suggests a role for the cochlea aqueduct in modulating distribution. Front. Cell Neurosci. 2019, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Choi, J.W.; Ishibashi, Y.; Isgrig, K.; Grati, M.; Bennett, J.; Chien, W. Refining surgical techniques for efficient posterior semicircular canal gene delivery in the adult mammalian inner ear with minimal hearing loss. Sci. Rep. 2021, 11, 18856. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 2017, 7, 45524. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.W.; Isgrig, K.; Roy, S.; Belyantseva, I.A.; Drummond, M.C.; May, L.A.; Fitzgerald, T.S.; Friedman, T.B.; Cunningham, L.L. Gene therapy restores hair cell stereocilia morphology in inner ears of deaf whirler mice. Mol. Ther. 2016, 24, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther. 2011, 18, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Tao, Y.; Wang, Z.; Tang, Y.; Li, H.; Dai, P.; Gao, G.; Chen, Z.Y. Identification of adeno-associated viral vectors that target neonatal and adult mammalian inner ear cell subtypes. Hum. Gene Ther. 2016, 27, 687–699. [Google Scholar] [CrossRef]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear gene transfer mediated by adeno-associated virus: Comparison of two surgical approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nist-Lund, C.; Solanes, P.; Goldberg, H.; Wu, J.; Pan, B.; Schneider, B.L.; Holt, J.R. Efficient viral transduction in mouse inner ear hair cells with utricle injection and AAV9-PHP.B. Hear. Res. 2020, 394, 107882. [Google Scholar] [CrossRef]
- Dror, A.A.; Avraham, K.B. Hearing loss: Mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 2009, 43, 411–437. [Google Scholar] [CrossRef]
- Wang, L.; Kempton, J.B.; Brigande, J.V. Gene therapy in mouse models of deafness and balance dysfunction. Front. Mol. Neurosci. 2018, 11, 300. [Google Scholar] [CrossRef]
- Litovsky, R. Development of the auditory system. Handb. Clin. Neurol. 2015, 129, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Brichta, A.M. Anatomical and physiological development of the human inner ear. Hear. Res. 2016, 338, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; et al. Delivery of adeno-associated virus vectors in adult mammalian inner-ear cell subtypes without auditory dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef]
- Zheng, Q.Y.; Johnson, K.R.; Erway, L.C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear. Res. 1999, 130, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Kane, K.L.; Longo-Guess, C.M.; Gagnon, L.H.; Ding, D.; Salvi, R.J.; Johnson, K.R. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hear. Res. 2012, 283, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Tian, C.; Gagnon, L.H.; Jiang, H.; Ding, D.; Salvi, R. Effects of Cdh23 single nucleotide substitutions on age-related hearing loss in C57BL/6 and 129S1/Sv mice and comparisons with congenic strains. Sci. Rep. 2017, 7, 44450. [Google Scholar] [CrossRef] [PubMed]
- Mianne, J.; Chessum, L.; Kumar, S.; Aguilar, C.; Codner, G.; Hutchison, M.; Parker, A.; Mallon, A.M.; Wells, S.; Simon, M.M.; et al. Correction of the auditory phenotype in C57BL/6N mice via CRISPR/Cas9-mediated homology directed repair. Genome Med. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Arjomandnejad, M.; Dasgupta, I.; Flotte, T.R.; Keeler, A.M. Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs 2023, 37, 311–329. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Sung, C.Y.W.; Grati, M.; Chien, W. Immune responses in the mammalian inner ear and their implications for AAV-mediated inner ear gene therapy. Hear. Res. 2023, 432, 108735. [Google Scholar] [CrossRef]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 can induce innate and adaptive immune response in the primate eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Gene Therapy Trial for Otoferlin Gene-Mediated Hearing Loss. Available online: https://classic.clinicaltrials.gov/show/NCT05821959 (accessed on 15 September 2023).
- A Study of DB-OTO, an AAV Based Gene Therapy, in Children/Infants with Hearing Loss Due to Otoferlin Mutations. Available online: https://classic.clinicaltrials.gov/show/NCT05788536 (accessed on 12 May 2023).


| Gene (Deafness Form) | Animal Model | Hearing Impairment | Injection Age | Injection Route | Vector | Pubmed |
|---|---|---|---|---|---|---|
| CLRN1 (USH3A) | TgAC1/Clrn1 KO | Delayed onset progressive | P1 | RWM | AAV-S | [18] |
| Clrn1 KO | Profound | P1–P3 | RWM | AAV2, AAV8 | [19] | |
| Clrn1e×4−/−/Clrn1 KO | Profound | P1–P3 | RWM | AAV8 | [20] | |
| Clrn1 KO | Profound | P1 | RWM | AAV9-PHP.B | [21] | |
| GJB2 (DFNA3A/DFNB1A) | Gjb2 iCKO | Severe to profound | P28 | RWM | Anc80L65 | [22] |
| Gjb2 iCKO | Profound | P0, P42 | RWM | AAV1 | [23] | |
| KCNE1 (JLNS2) | Kcne1 KO | Severe (balance defect) | P0–P2 | PSCC | AAV1 | [24] |
| KCNQ1 (JLNS1) | Kcnq1 KO | Severe (balance defect) | P0–P2 | RWM, Scala media | AAV1 | [25] |
| LHFPL5 (DFNB66/67) | Lhfpl5 KO | Profound (balance defect) | P1–P2 | RWM, Scala media | exo-AAV1 | [26] |
| MSRB3 (DFNB74) | Msrb3 KO | Profound | E12.5 | EUGO | AAV1 | [27] |
| OTOF (DFNB9) | Otof KO | Profound | P0–P2 | RWM | Dual-vector: AAV9-PHP.eB | [28] |
| Otof KO | Profound | P10, P17, P30 | RWM | Dual vector: AAV2 quadY-F | [29] | |
| Otof KO | Profound | P6–P7 | RWM | Dual vector: AAV6 | [30] | |
| PCDH15 (DFNB23/USH1F) | Pcdh15 KO | Profound (balance defect) | P1 | RWM | AAV9-PHP.B | [31] |
| PJVK (DFNB59) | Pjvk G292R/G292R | Progressive (balance defect) | P0–P1 | RWM | Anc80L65 | [32] |
| Pjvk KO | Variable (moderate to profound) | P3 | RWM | AAV8 | [33] | |
| SLC17A8 (DFNA25) | VGlut3 KO | Profound | 6–12 weeks | Cisterna magna | AAV9-PHP.B | [34] |
| VGlut3 KO | Profound | 5, 8, and 20 weeks | PSCC | AAV8 | [35] | |
| VGlut3 KO | Profound | P1–P3 and P10–P12 | RWM | AAV1 | [36] | |
| SLC26A4 (DFNB4) | Slc26a4 KO | Profound (balance defect) | E12.5 | EUGO | AAV1 | [37] |
| STRC (DFNB16) | Strc KO | Severe | P0–P1 | Utricle | Dual vector: AAV9-PHP.B | [38] |
| SYNE4 (DFNB76) | Syne4 KO | Severe to profound progressive | P0–P1.5 | PSCC | AAV2-9.PHP.B | [39] |
| TMC1 (DFNB7/11) | Tmc1 KO, Tmc1 Baringo | Profound | P1, P7 | Utricle | AAV9-PHP.B | [40] |
| Tmc1 KO, Tmc1 N1931/N1931 | Profound | P1 | Utricle | AAV2-9-PHP.B | [41] | |
| Tmc1 KO | Profound | P1–P2 | RWM | Anc80L65 | [42] | |
| Tmc1 KO, Tmc1 Bth | Profound | P0–P2 | RWM | AAV1 | [43] | |
| USH1C (DFNB18/USH1C) | Ush1c c.216G>A | Severe (balance defect) | P0–P1; P10–P12 | RWM | Anc80L65 | [44] |
| USH1G (USH1G) | Ush1g KO | Profound (balance defect) | P2.5 | RWM | AAV8 | [45] |
| WHRN (DFNB31) | Whrn wi/wi | Profound (balance defect) | P1–P5 | PSCC | AAV8 | [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, R.; Avraham, K.B. Gene Therapy for Inherited Hearing Loss: Updates and Remaining Challenges. Audiol. Res. 2023, 13, 952-966. https://doi.org/10.3390/audiolres13060083
Hahn R, Avraham KB. Gene Therapy for Inherited Hearing Loss: Updates and Remaining Challenges. Audiology Research. 2023; 13(6):952-966. https://doi.org/10.3390/audiolres13060083
Chicago/Turabian StyleHahn, Roni, and Karen B. Avraham. 2023. "Gene Therapy for Inherited Hearing Loss: Updates and Remaining Challenges" Audiology Research 13, no. 6: 952-966. https://doi.org/10.3390/audiolres13060083