
Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review



Abstract
:1. Introduction
2. Anatomy, Embryology, and Physiology of the Inner Ear
2.1. Principles
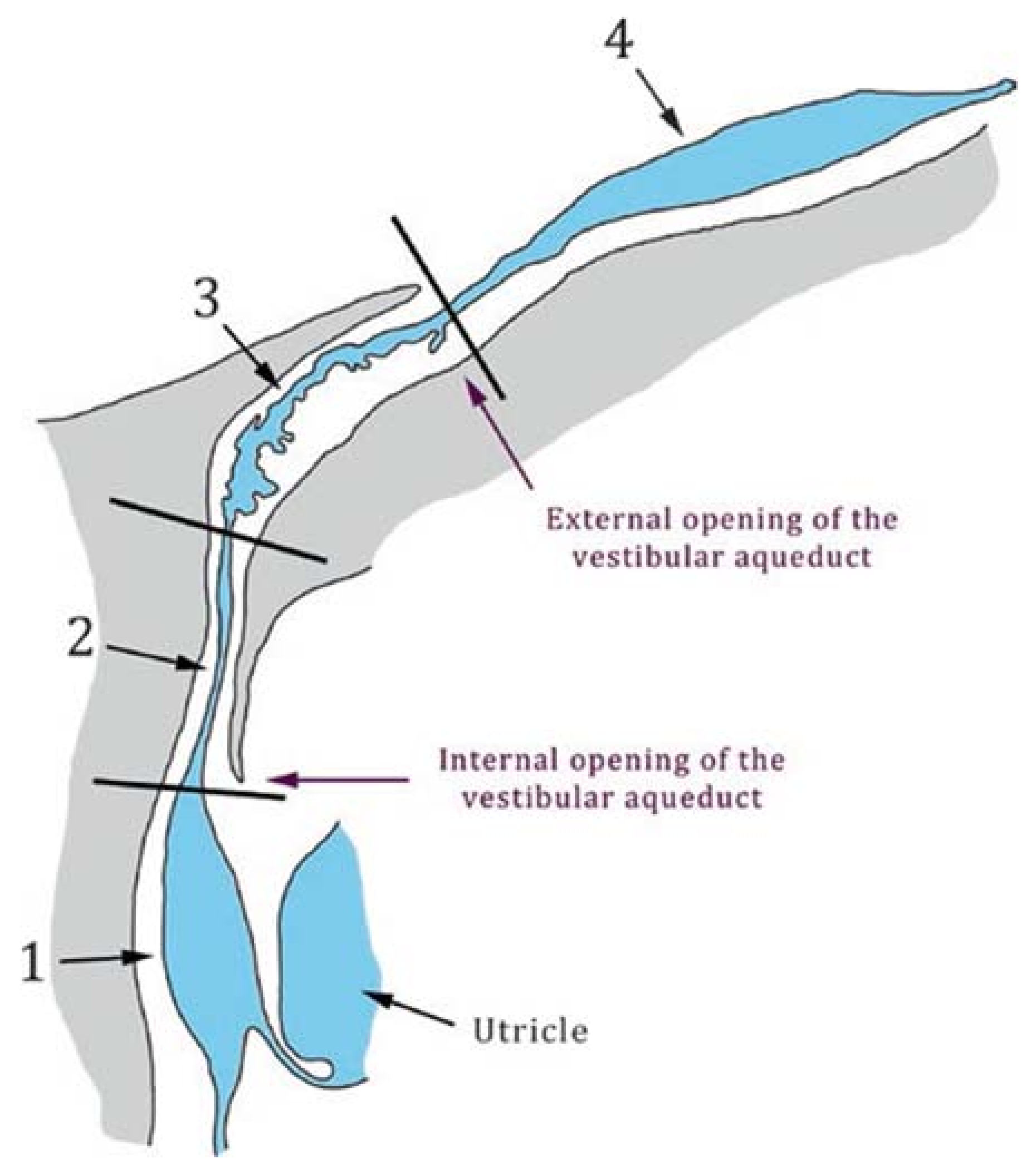
2.2. Anatomy of the Vestibular Aqueduct
3. Malformations of the Inner Ear
3.1. Classification
- Complete labyrinthine aplasia (Michel deformity): consists of the absence of the cochlea, vestibule, semicircular canals, vestibular and cochlear aqueduct.
- Cochlear aplasia: consists of the absence of the cochlea.
- Common cavity: consists of a single chamber representing the cochlea and vestibule.
- Rudimentary otocyst: represents an anomaly between a Michel deformity and common cavity.
- Cochlear hypoplasia: the cochlea is smaller than normal, with various internal architecture deformities.
- Incomplete partition type I (IP-I), or cystic cochleovestibular malformation: the cochlea lacks the entire modiolus and interscalar septa, and therefore has a cystic appearance.
- Incomplete partition type II (IP-II): the cochlea lacks the apical part of the modiolus. This anomaly was originally described by Carlo Mondini and together with a minimally dilated vestibule and an enlarged vestibular aqueduct (EVA) constitutes the triad of the Mondini deformity.
- Incomplete partition type III (IP-III): the cochlea has interscalar septa but the modiolus is completely absent. An IP-III is pathognomonic of X-linked deafness DFN3/DFNX2.
- Enlarged vestibular aqueduct (EVA): this is the most commonly detected malformation of the inner ear [24]. EVA is occasionally associated to other cochleovestibular malformations, such as cochlear incomplete partitions II or III, and can be found in syndromic as well as non-syndromic forms of hearing loss.
- Cochlear aperture abnormalities: the cochlear aperture transmits the cochlear nerve from the cochlea to the internal auditory canal. An aplastic or hypoplastic cochlear aperture is frequently associated to malfunction of the cochlear nerve.
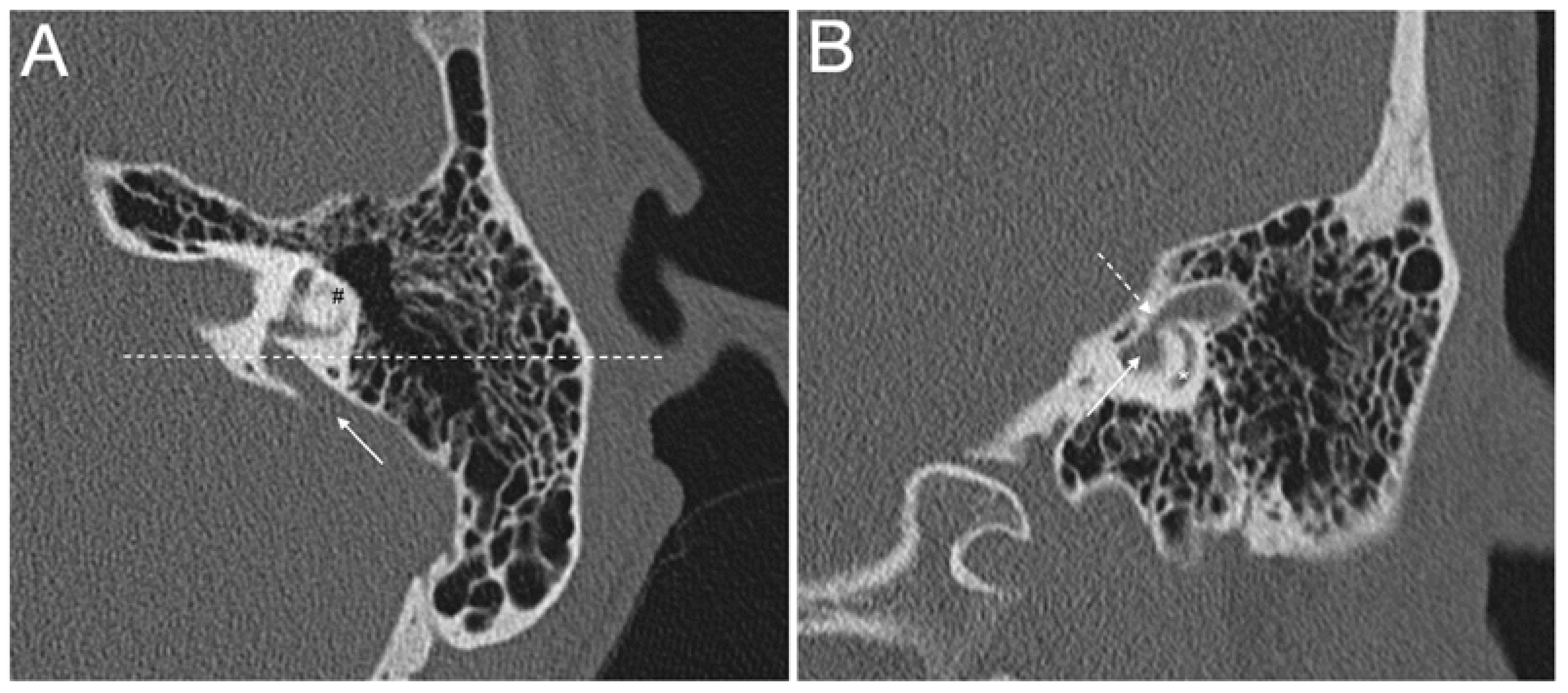
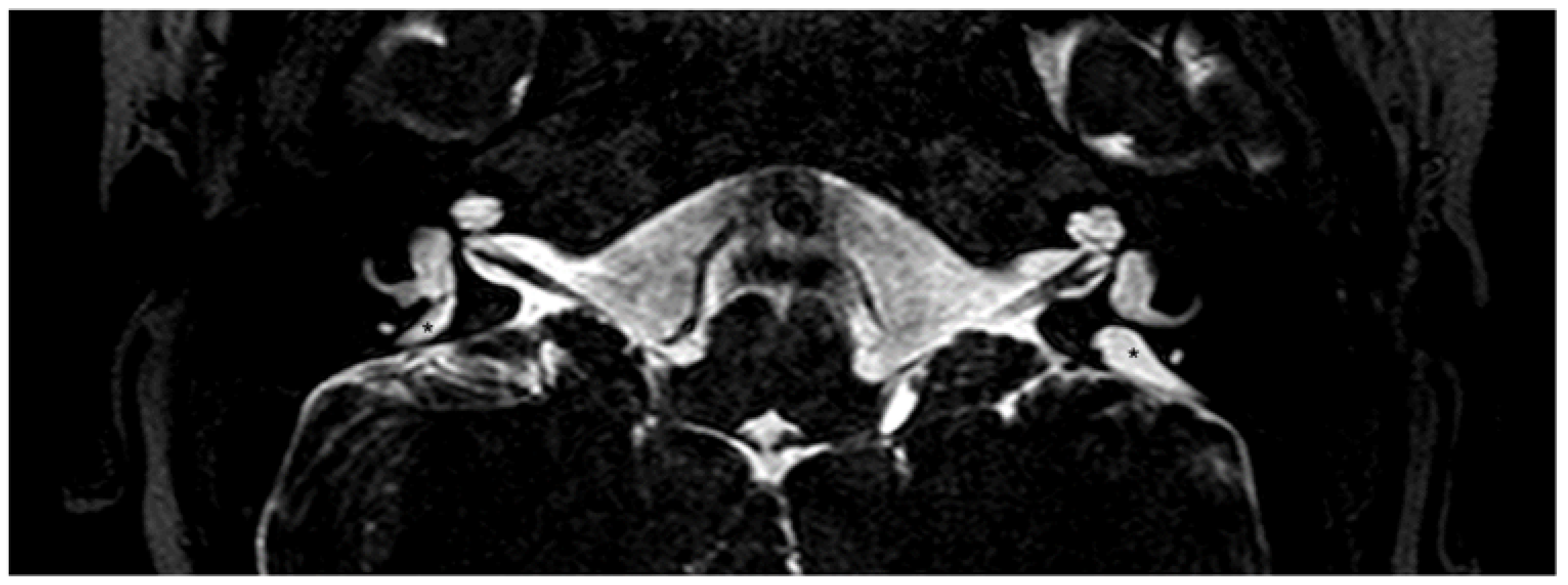
3.2. The Enlarged Vestibular Aqueduct
3.2.1. Definitions of Enlarged Vestibular Aqueduct
3.2.2. Diagnostic Tools for Enlarged Vestibular Aqueduct
4. Genes Involved in Determining Non-Syndromic Enlarged Vestibular Aqueduct
4.1. SLC26A4/Pendrin
4.1.1. DFNB4 and Pendred Syndrome
4.1.2. The SLC26A4/Pendrin Gene and Protein
4.1.3. Prevalence of SLC26A4 Sequence Alterations in Cohorts with EVA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probands (n) | M2 | M1 | M0 | Reference |
|---|---|---|---|---|
| 58 | 9 (16%) | 14 (24%) | 35 (60%) | [79] |
| 39 | 14 (36%) | 14 (36%) | 11 (28%) | [80] |
| 100 | 24 (24%) | 16 (16%) | 60 (60%) | [81] |
| 429 | 57 (13%) | 75 (17%) | 297 (69%) | [82] |
| 474 1 | 66 (14.5%) | 89 (19.5%) | 303 (66%) | [83] |
| 83 | 20 (24%) | 16 (19%) | 47 (57%) | [75] |
| 85 | 7 (8%) | 17 (20%) | 61 (72%) | [84] |
| 123 2 | 32 (26%) | 15 (12%) | 76 (62%) | [72] |
| 115 | 87 (76%) | 16 (14%) | 12 (10%) | [85] |
| 66 | 40.2% | 17.5% | 42.1 | [86] |
| Average | 28% | 19.5% | 52.5% |
4.1.4. SLC26A4 and Meniere Disease
4.2. Genes and Genetic Factors That Have Been Linked to EVA in Association with Monoallelic Pendrin Mutations
4.2.1. FOXI1 and KCNJ10
4.2.2. The Caucasian EVA (CEVA) Haplotype
4.2.3. EPHA2
4.3. POU3F4
4.4. GJB2
4.5. TMC1
4.6. Other Genetic Factors
5. Conclusions
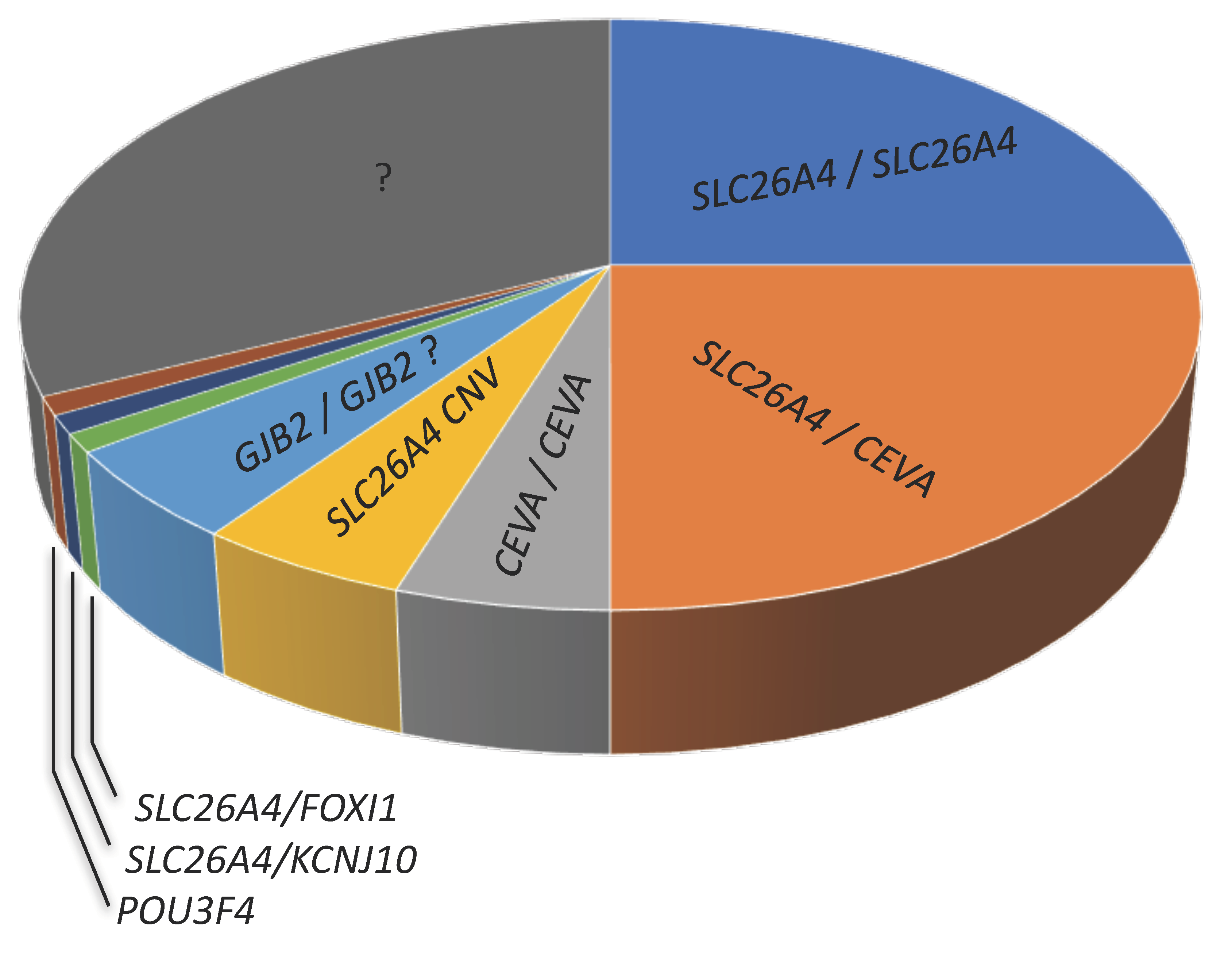
- In Caucasian cohorts, SLC26A4 and/or the CEVA haplotype, FOXI1, KCNJ10, POU3F4, and possibly GJB2 may account for hearing loss and EVA in approximately 50–65% of patients;
- The genes that are causative of EVA remain unidentified in 35–50% of patients (Figure 4);
- The genetic determinants of EVA in undiagnosed patients are probably strongly heterogeneous, especially in cases of unilateral EVA;
- In undiagnosed patients, causative sequence alterations may fall in regulatory regions or in genes coding for unidentified regulatory factors of the established genes and proteins, in genes not formerly linked to EVA, or even in genes not formerly linked to deafness.
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss: Syndromic. Otolaryngol. Clin. N. Am. 2015, 48, 1041–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, A. Genetic hearing loss: The journey of discovery to destination—How close are we to therapy? Mol. Genet. Genom. Med. 2016, 4, 583–587. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol Med. 2009, 9, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Kemperman, M.H.; Hoefsloot, L.H.; Cremers, C.W. Hearing loss and connexin 26. J. R. Soc. Med. 2002, 95, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Costa, S.M.; Ramos, P.Z.; Arrojo Martins, F.T.; Sartorato, E.L. Genetic Diagnosis of Deafness. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 61–83. [Google Scholar]
- Gonzalez-Garcia, J.A.; Ibanez, A.; Ramirez-Camacho, R.; Rodriguez, A.; Garcia-Berrocal, J.R.; Trinidad, A. Enlarged vestibular aqueduct: Looking for genotypic-phenotypic correlations. Eur. Arch. Otorhinolaryngol. 2006, 263, 971–976. [Google Scholar] [CrossRef]
- Tekin, M.; Sirmaci, A.; Yuksel-Konuk, B.; Fitoz, S.; Sennaroglu, L. A complex TFAP2A allele is associated with branchio-oculo-facial syndrome and inner ear malformation in a deaf child. Am. J. Med. Genet. Part A 2009, 149, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Bast, T.H.; Anson, B.J. The Temporal Bone and the Ear; Charles C Thomas Publisher: Springfield, IL, USA, 1949. [Google Scholar]
- Helwany, M.; Tadi, P. Embryology, Ear. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Scheuer, L.; Black, S. CHAPTER FIVE—The Head, Neck and Dentition. In Developmental Juvenile Osteology; Academic Press: Cambridge, MA, USA, 2000; pp. 36–170. [Google Scholar] [CrossRef]
- Tóth, M.; Csillag, A. The Organ of Hearing and Equilibrium. In Atlas of the Sensory Organs; Csillag, A., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 1–83. [Google Scholar] [CrossRef]
- Lundquist, P.G. Aspects on endolymphatic sac morphology and function. Arch. Otorhinolaryngol. 1976, 212, 231–240. [Google Scholar] [CrossRef]
- Couloigner, V.; Teixeira, M.; Sterkers, O.; Rask-Andersen, H.; Ferrary, E. The endolymphatic sac: Its roles in the inner ear. Med. Sci. (Paris) 2004, 20, 304–310. [Google Scholar] [CrossRef]
- Dahlmann, A.; von During, M. The endolymphatic duct and sac of the rat: A histological, ultrastructural, and immunocytochemical investigation. Cell Tissue Res. 1995, 282, 277–289. [Google Scholar] [CrossRef]
- Mori, N.; Miyashita, T.; Inamoto, R.; Matsubara, A.; Mori, T.; Akiyama, K.; Hoshikawa, H. Ion transport its regulation in the endolymphatic sac: Suggestions for clinical aspects of Meniere’s disease. Eur. Arch. Otorhinolaryngol. 2017, 274, 1813–1820. [Google Scholar] [CrossRef] [Green Version]
- Roesch, S.; Tóth, M.; Rasp, G. Pendrin-Linked Deafness in Humans. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 37–60. [Google Scholar]
- Lyu, H.; Hong, J.; Yin, D.; Chen, K.; Li, J.; Yang, L.; Zhang, T.; Dai, P. Position of the internal aperture of vestibular aqueduct in patients with enlarged vestibular aqueduct. Otol. Neurotol. 2017, 38, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Kämpfe Nordström, C. The Human Vestibular Aqueduct, Endolymphatic Duct and Sac: A Morphological Study Using Micro-ct, Super Resolution Immunohistochemistry and Synchrotron Phase Contrast Imaging. Ph.D. Thesis, Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine, Uppsala University, Uppsala, Sweden, 2020. [Google Scholar]
- Juliano, A.F.; Ting, E.Y.; Mingkwansook, V.; Hamberg, L.M.; Curtin, H.D. Vestibular aqueduct measurements in the 45 degrees oblique (Poschl) plane. AJNR Am. J. Neuroradiol. 2016, 37, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulos, B.; Zafar Gondal, A. Embryology, Ear Congenital Malformations. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Sennaroglu, L.; Bajin, M.D. Classification and current management of inner ear malformations. Balk. Med. J. 2017, 34, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L. Histopathology of inner ear malformations: Do we have enough evidence to explain pathophysiology? Cochlear Implant. Int. 2016, 17, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Usami, S.; Abe, S.; Weston, M.D.; Shinkawa, H.; Van Camp, G.; Kimberling, W.J. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum. Genet. 1999, 104, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, G.E.; Clemis, J.D. The large vestibular aqueduct syndrome. Laryngoscope 1978, 88, 723–728. [Google Scholar] [CrossRef]
- Vijayasekaran, S.; Halsted, M.J.; Boston, M.; Meinzen-Derr, J.; Bardo, D.M.; Greinwald, J.; Benton, C. When is the vestibular aqueduct enlarged? A statistical analysis of the normative distribution of vestibular aqueduct size. AJNR Am. J. Neuroradiol. 2007, 28, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Boston, M.; Halsted, M.; Meinzen-Derr, J.; Bean, J.; Vijayasekaran, S.; Arjmand, E.; Choo, D.; Benton, C.; Greinwald, J. The large vestibular aqueduct: A new definition based on audiologic and computed tomography correlation. Otolaryngol. Head Neck Surg. 2007, 136, 972–977. [Google Scholar] [CrossRef]
- Dewan, K.; Wippold, F.J., 2nd; Lieu, J.E. Enlarged vestibular aqueduct in pediatric sensorineural hearing loss. Otolaryngol. Head Neck Surg. 2009, 140, 552–558. [Google Scholar] [CrossRef]
- Muskett, J.A.; Chattaraj, P.; Heneghan, J.F.; Reimold, F.R.; Shmukler, B.E.; Brewer, C.C.; King, K.A.; Zalewski, C.K.; Shawker, T.H.; Butman, J.A.; et al. Atypical patterns of segregation of familial enlargement of the vestibular aqueduct. Laryngoscope 2016, 126, E240–E247. [Google Scholar] [CrossRef] [Green Version]
- Mafong, D.D.; Shin, E.J.; Lalwani, A.K. Use of laboratory evaluation and radiologic imaging in the diagnostic evaluation of children with sensorineural hearing loss. Laryngoscope 2002, 112, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sone, M.; Yoshida, T.; Morimoto, K.; Teranishi, M.; Nakashima, T.; Naganawa, S. Endolymphatic hydrops in superior canal dehiscence and large vestibular aqueduct syndromes. Laryngoscope 2016, 126, 1446–1450. [Google Scholar] [CrossRef]
- Tsukada, K.; Usami, S.I. Detailed MR imaging assessment of endolymphatic hydrops in patients with SLC26A4 mutations. Auris Nasus Larynx 2020, 47, 958–964. [Google Scholar] [CrossRef]
- Pendred, V. Deaf-mutism and goiter. Lancet 1896, 2, 532. [Google Scholar] [CrossRef] [Green Version]
- Wemeau, J.L.; Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 213–224. [Google Scholar] [CrossRef]
- Fugazzola, L.; Cirello, V.; Dossena, S.; Rodighiero, S.; Muzza, M.; Castorina, P.; Lalatta, F.; Ambrosetti, U.; Beck-Peccoz, P.; Botta, G.; et al. High phenotypic intrafamilial variability in patients with Pendred syndrome and a novel duplication in the SLC26A4 gene: Clinical characterization and functional studies of the mutated SLC26A4 protein. Eur. J. Endocrinol. 2007, 157, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Fraser, G.R. Association of congenital deafness with goitre (Pendred’s Syndrome) a study of 207 families. Ann. Hum. Genet. 1965, 28, 201–249. [Google Scholar] [CrossRef]
- Fugazzola, L.; Cerutti, N.; Mannavola, D.; Vannucchi, G.; Beck-Peccoz, P. The role of pendrin in iodide regulation. Exp. Clin. Endocrinol. Diabetes 2001, 109, 18–22. [Google Scholar] [CrossRef]
- Griffith, A.J.; Wangemann, P. Hearing loss associated with enlargement of the vestibular aqueduct: Mechanistic insights from clinical phenotypes, genotypes, and mouse models. Hear. Res. 2011, 281, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.J.H.; Iwasa, Y.; Schaefer, A.M. Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Choi, B.Y.; Kim, H.M.; Ito, T.; Lee, K.Y.; Li, X.; Monahan, K.; Wen, Y.; Wilson, E.; Kurima, K.; Saunders, T.L.; et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J. Clin. Investig. 2011, 121, 4516–4525. [Google Scholar] [CrossRef]
- Sennaroglu, L. Another evidence for pressure transfer mechanism in incomplete partition two anomaly via enlarged vestibular aqueduct. Cochlear Implant. Int. 2018, 19, 355–357. [Google Scholar] [CrossRef]
- Sennaroglu, L. Response to letter regarding ‘Another evidence for pressure transfer mechanism in incomplete partition two anomaly via enlarged vestibular aqueduct’. Cochlear Implant. Int. 2021, 22, 183–185. [Google Scholar] [CrossRef]
- Roesch, S.; Bernardinelli, E.; Nofziger, C.; Toth, M.; Patsch, W.; Rasp, G.; Paulmichl, M.; Dossena, S. Functional testing of SLC26A4 variants-clinical and molecular analysis of a cohort with enlarged vestibular aqueduct from Austria. Int. J. Mol. Sci. 2018, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Everett, L.A.; Glaser, B.; Beck, J.C.; Idol, J.R.; Buchs, A.; Heyman, M.; Adawi, F.; Hazani, E.; Nassir, E.; Baxevanis, A.D.; et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 1997, 17, 411–422. [Google Scholar] [CrossRef]
- Haila, S.; Hoglund, P.; Scherer, S.W.; Lee, J.R.; Kristo, P.; Coyle, B.; Trembath, R.; Holmberg, C.; de la Chapelle, A.; Kere, J. Genomic structure of the human congenital chloride diarrhea (CLD) gene. Gene 1998, 214, 87–93. [Google Scholar] [CrossRef]
- Everett, L.A.; Morsli, H.; Wu, D.K.; Green, E.D. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 1999, 96, 9727–9732. [Google Scholar] [CrossRef] [Green Version]
- Royaux, I.E.; Wall, S.M.; Karniski, L.P.; Everett, L.A.; Suzuki, K.; Knepper, M.A.; Green, E.D. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc. Natl. Acad. Sci. USA 2001, 98, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Wangemann, P.; Itza, E.M.; Albrecht, B.; Wu, T.; Jabba, S.V.; Maganti, R.J.; Lee, J.H.; Everett, L.A.; Wall, S.M.; Royaux, I.E.; et al. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004, 2, 30. [Google Scholar] [CrossRef]
- Wangemann, P.; Griffith, A.J. Mouse models reveal the role of pendrin in the inner ear. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 7–22. [Google Scholar]
- Korrapati, S.; Taukulis, I.; Olszewski, R.; Pyle, M.; Gu, S.; Singh, R.; Griffiths, C.; Martin, D.; Boger, E.; Morell, R.J.; et al. Single cell and single nucleus RNA-Seq reveal cellular heterogeneity and homeostatic regulatory networks in adult mouse stria vascularis. Front. Mol. Neurosci. 2019, 12, 316. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Wangemann, P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS ONE 2011, 6, e17949. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, K.; Harbidge, D.G.; Wangemann, P.; Schultz, B.D.; Green, E.D.; Wall, S.M.; Marcus, D.C. Lack of pendrin HCO3− transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am. J. Physiol. Ren. Physiol. 2007, 292, F1314–F1321. [Google Scholar] [CrossRef] [Green Version]
- Soleimani, M. The multiple roles of pendrin in the kidney. Nephrol. Dial. Transplant. 2015, 30, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Nishio, A.; Ito, T.; Cheng, H.; Fitzgerald, T.S.; Wangemann, P.; Griffith, A.J. Slc26a4 expression prevents fluctuation of hearing in a mouse model of large vestibular aqueduct syndrome. Neuroscience 2016, 329, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Wangemann, P. Mouse models for pendrin-associated loss of cochlear and vestibular function. Cell Physiol. Biochem. 2013, 32, 157–165. [Google Scholar] [CrossRef]
- Wen, Z.; Zhu, H.; Li, Z.; Zhang, S.; Zhang, A.; Zhang, T.; Fu, X.; Sun, D.; Zhang, J.; Gao, J. A knock-in mouse model of Pendred syndrome with Slc26a4 L236P mutation. Biochem. Biophys. Res. Commun. 2019, 515, 359–365. [Google Scholar] [CrossRef]
- Takeda, H.; Miwa, T.; Kim, M.Y.; Choi, B.Y.; Orita, Y.; Minoda, R. Prenatal electroporation-mediated gene transfer restores Slc26a4 knock-out mouse hearing and vestibular function. Sci. Rep. 2019, 9, 17979. [Google Scholar] [CrossRef]
- Taylor, J.P.; Metcalfe, R.A.; Watson, P.F.; Weetman, A.P.; Trembath, R.C. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: Implications for thyroid dysfunction in Pendred syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 1778–1784. [Google Scholar] [CrossRef]
- Rotman-Pikielny, P.; Hirschberg, K.; Maruvada, P.; Suzuki, K.; Royaux, I.E.; Green, E.D.; Kohn, L.D.; Lippincott-Schwartz, J.; Yen, P.M. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum. Mol. Genet. 2002, 11, 2625–2633. [Google Scholar] [CrossRef] [Green Version]
- De Moraes, V.C.; Bernardinelli, E.; Zocal, N.; Fernandez, J.A.; Nofziger, C.; Castilho, A.M.; Sartorato, E.L.; Paulmichl, M.; Dossena, S. Reduction of cellular expression levels is a common feature of functionally affected pendrin (SLC26A4) protein variants. Mol. Med. 2016, 22, 41–53. [Google Scholar] [CrossRef]
- Dror, A.A.; Politi, Y.; Shahin, H.; Lenz, D.R.; Dossena, S.; Nofziger, C.; Fuchs, H.; Hrabe de Angelis, M.; Paulmichl, M.; Weiner, S.; et al. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J. Biol. Chem. 2010, 285, 21724–21735. [Google Scholar] [CrossRef] [Green Version]
- Dror, A.A.; Lenz, D.R.; Shivatzki, S.; Cohen, K.; Ashur-Fabian, O.; Avraham, K.B. Atrophic thyroid follicles and inner ear defects reminiscent of cochlear hypothyroidism in Slc26a4-related deafness. Mamm. Genome 2014, 25, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Liang, F.; Zhao, M.; Gong, A.; Berry, E.R.; Shi, Y.; Wang, Y.; Chen, Y.; Liu, A.; Qu, C. Mutational analysis of the SLC26A4 gene in Chinese sporadic nonsyndromic hearing-impaired children. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1474–1480. [Google Scholar] [CrossRef]
- Qing, J.; Zhou, Y.; Lai, R.; Hu, P.; Ding, Y.; Wu, W.; Xiao, Z.; Ho, P.T.; Liu, Y.; Liu, J.; et al. Prevalence of mutations in GJB2, SLC26A4, and mtDNA in children with severe or profound sensorineural hearing loss in southwestern China. Genet. Test. Mol. Biomark. 2015, 19, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Chai, Y.; Chen, P.; He, L.; Wang, X.; Wu, H.; Yang, T. Mono-allelic mutations of SLC26A4 is over-presented in deaf patients with non-syndromic enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1351–1353. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, H.; Liu, D.; Zhang, J.; Yang, Z.; Zhang, S.; Liu, H.; Li, R.; Tian, Y.; Zeng, B.; et al. Increased diagnosis of enlarged vestibular aqueduct by multiplex PCR enrichment and next-generation sequencing of the SLC26A4 gene. Mol. Genet. Genom. Med. 2021, e1734, Online ahead of print. [Google Scholar] [CrossRef]
- Lin, Y.H.; Wu, C.C.; Lin, Y.H.; Lu, Y.C.; Chen, C.S.; Liu, T.C.; Chen, P.L.; Hsu, C.J. Targeted next-generation sequencing facilitates genetic diagnosis and provides novel pathogenetic insights into deafness with enlarged vestibular aqueduct. J. Mol. Diagn. 2019, 21, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Lee, S.C.; Lee, H.K.; Park, H.J. Genetic screening of GJB2 and SLC26A4 in Korean cochlear implantees: Experience of Soree ear clinic. Clin. Exp. Otorhinolaryngol. 2012, 5 (Suppl. 1), S10–S13. [Google Scholar] [CrossRef]
- Rah, Y.C.; Kim, A.R.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef]
- Miyagawa, M.; Nishio, S.Y.; Usami, S.; Deafness Gene Study Consortium. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Muskett, J.; Chattaraj, P.; Choi, B.Y.; Lee, K.Y.; Zalewski, C.K.; King, K.A.; Li, X.; Wangemann, P.; Shawker, T.; et al. SLC26A4 mutation testing for hearing loss associated with enlargement of the vestibular aqueduct. World J. Otorhinolaryngol. 2013, 3, 26–34. [Google Scholar] [CrossRef]
- Rose, J.; Muskett, J.A.; King, K.A.; Zalewski, C.K.; Chattaraj, P.; Butman, J.A.; Kenna, M.A.; Chien, W.W.; Brewer, C.C.; Griffith, A.J. Hearing loss associated with enlarged vestibular aqueduct and zero or one mutant allele of SLC26A4. Laryngoscope 2017, 127, E238–E243. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Friedman, T.B.; Griffith, A.J. Unresolved questions regarding human hereditary deafness. Oral. Dis. 2017, 23, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Madeo, A.C.; King, K.A.; Zalewski, C.K.; Pryor, S.P.; Muskett, J.A.; Nance, W.E.; Butman, J.A.; Brewer, C.C.; Griffith, A.J. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J. Med. Genet. 2009, 46, 856–861. [Google Scholar] [CrossRef]
- King, K.A.; Choi, B.Y.; Zalewski, C.; Madeo, A.C.; Manichaikul, A.; Pryor, S.P.; Ferruggiaro, A.; Eisenman, D.; Kim, H.J.; Niparko, J.; et al. SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope 2010, 120, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Pique, L.M.; Brennan, M.L.; Davidson, C.J.; Schaefer, F.; Greinwald, J., Jr.; Schrijver, I. Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. Peer J. 2014, 2, e384. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Wang, L.L.; Wen, J.; Mei, L.Y.; He, C.F.; Jiang, L.; Feng, Y. Application value of high-throughput gene copy number variation detection in the diagnosis of enlarged vestibular aqueduct. Zhonghua Yi Xue Za Zhi 2021, 101, 103–107. [Google Scholar] [CrossRef]
- Chattaraj, P.; Reimold, F.R.; Muskett, J.A.; Shmukler, B.E.; Chien, W.W.; Madeo, A.C.; Pryor, S.P.; Zalewski, C.K.; Butman, J.A.; Brewer, C.C.; et al. Use of SLC26A4 mutation testing for unilateral enlargement of the vestibular aqueduct. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Campbell, C.; Cucci, R.A.; Prasad, S.; Green, G.E.; Edeal, J.B.; Galer, C.E.; Karniski, L.P.; Sheffield, V.C.; Smith, R.J. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum. Mutat. 2001, 17, 403–411. [Google Scholar] [CrossRef]
- Pryor, S.P.; Madeo, A.C.; Reynolds, J.C.; Sarlis, N.J.; Arnos, K.S.; Nance, W.E.; Yang, Y.; Zalewski, C.K.; Brewer, C.C.; Butman, J.A.; et al. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): Evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J. Med. Genet. 2005, 42, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.; Blons, H.; Jonard, L.; Feldmann, D.; Chauvin, P.; Loundon, N.; Sergent-Allaoui, A.; Houang, M.; Joannard, A.; Schmerber, S.; et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur. J. Hum. Genet. 2006, 14, 773–779. [Google Scholar] [CrossRef]
- Yang, T.; Vidarsson, H.; Rodrigo-Blomqvist, S.; Rosengren, S.S.; Enerback, S.; Smith, R.J. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am. J. Hum. Genet. 2007, 80, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azaiez, H.; Yang, T.; Prasad, S.; Sorensen, J.L.; Nishimura, C.J.; Kimberling, W.J.; Smith, R.J. Genotype-phenotype correlations for SLC26A4-related deafness. Hum. Genet. 2007, 122, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Greinwald, J.; DeAlarcon, A.; Cohen, A.; Uwiera, T.; Zhang, K.; Benton, C.; Halstead, M.; Meinzen-Derr, J. Significance of unilateral enlarged vestibular aqueduct. Laryngoscope 2013, 123, 1537–1546. [Google Scholar] [CrossRef]
- Mey, K.; Muhamad, A.A.; Tranebjaerg, L.; Rendtorff, N.D.; Rasmussen, S.H.; Bille, M.; Caye-Thomasen, P. Association of SLC26A4 mutations, morphology, and hearing in pendred syndrome and NSEVA. Laryngoscope 2019, 129, 2574–2579. [Google Scholar] [CrossRef]
- Forli, F.; Lazzerini, F.; Auletta, G.; Bruschini, L.; Berrettini, S. Enlarged vestibular aqueduct and Mondini malformation: Audiological, clinical, radiologic and genetic features. Eur. Arch. Otorhinolaryngol. 2021, 278, 2305–2312. [Google Scholar] [CrossRef]
- Merchant, S.N.; Adams, J.C.; Nadol, J.B., Jr. Pathophysiology of Meniere’s syndrome: Are symptoms caused by endolymphatic hydrops? Otol. Neurotol. 2005, 26, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.H.; Lalwani, A.K. Large vestibular aqueduct syndrome and endolymphatic hydrops: Two presentations of a common primary inner-ear dysfunction? J. Laryngol. Otol. 2009, 123, 919–921. [Google Scholar] [CrossRef]
- Gu, S.; Olszewski, R.; Nelson, L.; Gallego-Martinez, A.; Lopez-Escamez, J.A.; Hoa, M. Identification of potential Meniere’s disease targets in the adult stria vascularis. Front. Neurol. 2021, 12, 630561. [Google Scholar] [CrossRef]
- Gallego-Martinez, A.; Requena, T.; Roman-Naranjo, P.; Lopez-Escamez, J.A. Excess of rare missense variants in hearing loss genes in sporadic Meniere disease. Front. Genet. 2019, 10, 76. [Google Scholar] [CrossRef]
- Hulander, M.; Kiernan, A.E.; Blomqvist, S.R.; Carlsson, P.; Samuelsson, E.J.; Johansson, B.R.; Steel, K.P.; Enerback, S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development 2003, 130, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Marcus, D.C.; Wu, T.; Wangemann, P.; Kofuji, P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell Physiol. 2002, 282, C403–C407. [Google Scholar] [CrossRef] [Green Version]
- Enerback, S.; Nilsson, D.; Edwards, N.; Heglind, M.; Alkanderi, S.; Ashton, E.; Deeb, A.; Kokash, F.E.B.; Bakhsh, A.R.A.; Van’t Hoff, W.; et al. Acidosis and deafness in patients with recessive mutations in FOXI1. J. Am. Soc. Nephrol. 2018, 29, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, H.; Westergren, R.; Heglind, M.; Blomqvist, S.R.; Breton, S.; Enerback, S. The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS ONE 2009, 4, e4471. [Google Scholar] [CrossRef]
- Blomqvist, S.R.; Vidarsson, H.; Fitzgerald, S.; Johansson, B.R.; Ollerstam, A.; Brown, R.; Persson, A.E.; Bergstrom, G.G.; Enerback, S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J. Clin. Investig. 2004, 113, 1560–1570. [Google Scholar] [CrossRef] [Green Version]
- Berrettini, S.; Forli, F.; Franceschini, S.S.; Ravecca, F.; Massimetti, M.; Neri, E. Distal renal tubular acidosis associated with isolated large vestibular aqueduct and sensorineural hearing loss. Ann. Otol. Rhinol. Laryngol. 2002, 111, 385–391. [Google Scholar] [CrossRef]
- Andreucci, E.; Bianchi, B.; Carboni, I.; Lavoratti, G.; Mortilla, M.; Fonda, C.; Bigozzi, M.; Genuardi, M.; Giglio, S.; Pela, I. Inner ear abnormalities in four patients with dRTA and SNHL: Clinical and genetic heterogeneity. Pediatr. Nephrol. 2009, 24, 2147–2153. [Google Scholar] [CrossRef]
- Yashima, T.; Noguchi, Y.; Kawashima, Y.; Rai, T.; Ito, T.; Kitamura, K. Novel ATP6V1B1 mutations in distal renal tubular acidosis and hearing loss. Acta Otolaryngol. 2010, 130, 1002–1008. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, Y.; Li, Q.; Lang, Y.; Dong, Q.; Shao, L. Mutation analysis and audiologic assessment in six Chinese children with primary distal renal tubular acidosis. Ren. Fail. 2014, 36, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Lu, J.; Gao, Y.; Wang, X.; Lang, Y.; Shao, L. Novel compound heterozygous ATP6V1B1 mutations in a Chinese child patient with primary distal renal tubular acidosis: A case report. BMC Nephrol. 2018, 19, 364. [Google Scholar] [CrossRef]
- Dahmani, M.; Talbi, S.; Ammar-Khodja, F.; Ouhab, S.; Boudjenah, F.; Djebbar, M.; Bonnet, C.; Petit, C. ATP6V1B1 recurrent mutations in Algerian deaf patients associated with renal tubular acidosis. Int. J. Pediatr. Otorhinolaryngol. 2020, 129, 109772. [Google Scholar] [CrossRef]
- Tian, C.; Gagnon, L.H.; Longo-Guess, C.; Korstanje, R.; Sheehan, S.M.; Ohlemiller, K.K.; Schrader, A.D.; Lett, J.M.; Johnson, K.R. Hearing loss without overt metabolic acidosis in ATP6V1B1 deficient MRL mice, a new genetic model for non-syndromic deafness with enlarged vestibular aqueducts. Hum. Mol. Genet. 2017, 26, 3722–3735. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Gurrola, J.G.; Wu, H.; Chiu, S.M.; Wangemann, P.; Snyder, P.M.; Smith, R.J. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am. J. Hum. Genet. 2009, 84, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landa, P.; Differ, A.M.; Rajput, K.; Jenkins, L.; Bitner-Glindzicz, M. Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med. Genet. 2013, 14, 85. [Google Scholar] [CrossRef] [Green Version]
- Cirello, V.; Bazzini, C.; Vezzoli, V.; Muzza, M.; Rodighiero, S.; Castorina, P.; Maffini, A.; Botta, G.; Persani, L.; Beck-Peccoz, P.; et al. Molecular and functional studies of 4 candidate loci in Pendred syndrome and nonsyndromic hearing loss. Mol. Cell. Endocrinol. 2012, 351, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yuan, Y.; Huang, S.; Huang, B.; Cheng, J.; Kang, D.; Wang, G.; Han, D.; Dai, P. KCNJ10 may not be a contributor to nonsyndromic enlargement of vestibular aqueduct (NSEVA) in Chinese subjects. PLoS ONE 2014, 9, e108134. [Google Scholar] [CrossRef] [Green Version]
- Jonard, L.; Niasme-Grare, M.; Bonnet, C.; Feldmann, D.; Rouillon, I.; Loundon, N.; Calais, C.; Catros, H.; David, A.; Dollfus, H.; et al. Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 1049–1053. [Google Scholar] [CrossRef]
- Mercer, S.; Mutton, P.; Dahl, H.H. Identification of SLC26A4 mutations in patients with hearing loss and enlarged vestibular aqueduct using high-resolution melting curve analysis. Genet. Test. Mol. Biomark. 2011, 15, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lu, Y.C.; Chen, P.J.; Yeh, P.L.; Su, Y.N.; Hwu, W.L.; Hsu, C.J. Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol. Neurootol. 2010, 15, 57–66. [Google Scholar] [CrossRef]
- Chai, Y.; Huang, Z.; Tao, Z.; Li, X.; Li, L.; Li, Y.; Wu, H.; Yang, T. Molecular etiology of hearing impairment associated with nonsyndromic enlarged vestibular aqueduct in East China. Am. J. Med. Genet. Part A 2013, 161, 2226–2233. [Google Scholar] [CrossRef]
- Chen, K.; Wang, X.; Sun, L.; Jiang, H. Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngol. Head Neck Surg. 2012, 146, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Hu, P.; Zhu, F.; Zhu, G.; Vivero, R.; Peng, A.; Wu, W.; Xiao, Z.; Liu, X.; Xie, D. Genetic diagnosis and cochlear implantation for patients with nonsyndromic hearing loss and enlarged vestibular aqueduct. J. Laryngol. Otol. 2012, 126, 349–355. [Google Scholar] [CrossRef]
- Song, M.H.; Shin, J.W.; Park, H.J.; Lee, K.A.; Kim, Y.; Kim, U.K.; Jeon, J.H.; Choi, J.Y. Intrafamilial phenotypic variability in families with biallelic SLC26A4 mutations. Laryngoscope 2014, 124, E194–E202. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Feng, Y.; He, C.; Liu, D.; Cai, X.; Jiang, L.; Chen, H.; Liu, C.; Wu, H.; et al. A new genetic diagnostic for enlarged vestibular aqueduct based on next-generation sequencing. PLoS ONE 2016, 11, e0168508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wen, J.; Sang, S.; Mei, L.; He, C.; Jiang, L.; Huang, S.; Feng, Y. Next-generation sequencing-based mutation analysis of genes associated with enlarged vestibular aqueduct in Chinese families. Eur. Arch. Otorhinolaryngol. 2020, 277, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Munjal, T.; Honda, K.; Rendtorff, N.D.; Ratay, J.S.; Muskett, J.A.; Risso, D.S.; Roux, I.; Gertz, E.M.; Schaffer, A.A.; et al. A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J. Med. Genet. 2017, 54, 665–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.R.; Chattaraj, P.; Munjal, T.; Honda, K.; King, K.A.; Zalewski, C.K.; Chien, W.W.; Brewer, C.C.; Griffith, A.J. SLC26A4-linked CEVA haplotype correlates with phenotype in patients with enlargement of the vestibular aqueduct. BMC Med. Genet. 2019, 20, 118. [Google Scholar] [CrossRef] [Green Version]
- Darling, T.K.; Lamb, T.J. Emerging roles for Eph receptors and Ephrin ligands in immunity. Front. Immunol. 2019, 10, 1473. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Nishio, S.Y.; Naruse, C.; Riddell, M.; Sapski, S.; Katsuno, T.; Hikita, T.; Mizapourshafiyi, F.; Smith, F.M.; Cooper, L.T.; et al. Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome. Nat. Commun. 2020, 11, 1343. [Google Scholar] [CrossRef]
- Mathis, J.M.; Simmons, D.M.; He, X.; Swanson, L.W.; Rosenfeld, M.G. Brain 4: A novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 1992, 11, 2551–2561. [Google Scholar] [CrossRef]
- de Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef] [Green Version]
- Minowa, O.; Ikeda, K.; Sugitani, Y.; Oshima, T.; Nakai, S.; Katori, Y.; Suzuki, M.; Furukawa, M.; Kawase, T.; Zheng, Y.; et al. Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science 1999, 285, 1408–1411. [Google Scholar] [CrossRef]
- Song, M.H.; Choi, S.Y.; Wu, L.; Oh, S.K.; Lee, H.K.; Lee, D.J.; Shim, D.B.; Choi, J.Y.; Kim, U.K.; Bok, J. Pou3f4 deficiency causes defects in otic fibrocytes and stria vascularis by different mechanisms. Biochem. Biophys. Res. Commun. 2011, 404, 528–533. [Google Scholar] [CrossRef]
- Singh, R.; Wangemann, P. Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am. J. Physiol. Renal. Physiol. 2008, 294, F139–F148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vore, A.P.; Chang, E.H.; Hoppe, J.E.; Butler, M.G.; Forrester, S.; Schneider, M.C.; Smith, L.L.; Burke, D.W.; Campbell, C.A.; Smith, R.J. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Chee, N.W.; Suhailee, S.; Goh, J. Clinics in diagnostic imaging (111): X-linked congenital mixed deafness syndrome. Singap. Med. J. 2006, 47, 822–824; quiz 825. [Google Scholar] [PubMed]
- Marlin, S.; Moizard, M.P.; David, A.; Chaissang, N.; Raynaud, M.; Jonard, L.; Feldmann, D.; Loundon, N.; Denoyelle, F.; Toutain, A. Phenotype and genotype in females with POU3F4 mutations. Clin. Genet. 2009, 76, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.X.; Gong, R.Z.; Zhao, B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; An, Y.H.; Park, J.H.; Jang, J.H.; Chung, H.C.; Kim, A.R.; Lee, J.H.; Kim, C.S.; Oh, S.H.; Chang, S.O. Audiological and surgical evidence for the presence of a third window effect for the conductive hearing loss in DFNX2 deafness irrespective of types of mutations. Eur. Arch. Otorhinolaryngol. 2013, 270, 3057–3062. [Google Scholar] [CrossRef]
- Pollak, A.; Lechowicz, U.; Kedra, A.; Stawinski, P.; Rydzanicz, M.; Furmanek, M.; Brzozowska, M.; Mrowka, M.; Skarzynski, H.; Skarzynski, P.H.; et al. Novel and de novo mutations extend association of POU3F4 with distinct clinical and radiological phenotype of hearing loss. PLoS ONE 2016, 11, e0166618. [Google Scholar] [CrossRef]
- Incesulu, A.; Adapinar, B.; Kecik, C. Cochlear implantation in cases with incomplete partition type III (X-linked anomaly). Eur. Arch. Otorhinolaryngol. 2008, 265, 1425–1430. [Google Scholar] [CrossRef]
- Kanno, A.; Mutai, H.; Namba, K.; Morita, N.; Nakano, A.; Ogahara, N.; Sugiuchi, T.; Ogawa, K.; Matsunaga, T. Frequency and specific characteristics of the incomplete partition type III anomaly in children. Laryngoscope 2017, 127, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.A.; Ozutemiz, C.; Miller, B.S.; Moss, T.J.; Nascene, D.R. Hypothalamic hamartomas and inner ear diverticula with X-linked stapes gusher syndrome—New associations? Pediatr. Radiol. 2020, 50, 142–145. [Google Scholar] [CrossRef]
- Kikuchi, T.; Kimura, R.S.; Paul, D.L.; Takasaka, T.; Adams, J.C. Gap junction systems in the mammalian cochlea. Brain Res. Rev. 2000, 32, 163–166. [Google Scholar] [CrossRef]
- Verselis, V.K. Connexin hemichannels and cochlear function. Neurosci. Lett. 2019, 695, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Schrijver, I.; Chang, K.W. Two patients with the V37I/235delC genotype: Are radiographic cochlear anomalies part of the phenotype? Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 2109–2113. [Google Scholar] [CrossRef]
- Santos, S.; Sgambatti, L.; Bueno, A.; Albi, G.; Suarez, A.; Dominguez, M.J. Enlarged vestibular aqueduct syndrome. A review of 55 paediatric patients. Acta Otorrinolaringol. Esp. 2010, 61, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Deklerck, A.N.; Acke, F.R.; Janssens, S.; De Leenheer, E.M. Etiological approach in patients with unidentified hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 216–222. [Google Scholar] [CrossRef]
- Xiang, Y.; Li, H.; Xu, X.; Xu, C.; Chen, C.; Lin, X.; Tang, S. Mutation analysis and prenatal diagnosis for 12 families affected with hereditary hearing loss and enlarged vestibular aqueduct. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2017, 34, 336–341. [Google Scholar] [CrossRef]
- Wu, H.; Jiang, L.; Liu, C.; Liu, Y.L.; Long, M.Q.; Mei, L.Y.; He, C.F.; Cai, X.Z.; Chen, H.S.; Feng, Y. Validation and analysis of Goldengate high-throughput deafness gene chip in detecting the patients with enlarged vestibular aqueduct syndrome. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2021, 56, 138–143. [Google Scholar] [CrossRef]
- Propst, E.J.; Blaser, S.; Stockley, T.L.; Harrison, R.V.; Gordon, K.A.; Papsin, B.C. Temporal bone imaging in GJB2 deafness. Laryngoscope 2006, 116, 2178–2186. [Google Scholar] [CrossRef]
- Lee, K.H.; Larson, D.A.; Shott, G.; Rasmussen, B.; Cohen, A.P.; Benton, C.; Halsted, M.; Choo, D.; Meinzen-Derr, J.; Greinwald, J.H., Jr. Audiologic and temporal bone imaging findings in patients with sensorineural hearing loss and GJB2 mutations. Laryngoscope 2009, 119, 554–558. [Google Scholar] [CrossRef]
- Kenna, M.A.; Rehm, H.L.; Frangulov, A.; Feldman, H.A.; Robson, C.D. Temporal bone abnormalities in children with GJB2 mutations. Laryngoscope 2011, 121, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Han, D.; Wang, G.; Yuan, Y.; Song, Y.; Han, M.; Chen, Z.; Dai, P. Sensorineural hearing loss caused by mutations in two alleles of both GJB2 and SLC26A4 genes. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 379–383. [Google Scholar] [CrossRef]
- Kurima, K.; Peters, L.M.; Yang, Y.; Riazuddin, S.; Ahmed, Z.M.; Naz, S.; Arnaud, D.; Drury, S.; Mo, J.; Makishima, T.; et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat. Genet. 2002, 30, 277–284. [Google Scholar] [CrossRef]
- Nakanishi, H.; Kurima, K.; Kawashima, Y.; Griffith, A.J. Mutations of TMC1 cause deafness by disrupting mechanoelectrical transduction. Auris Nasus Larynx 2014, 41, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohne, A.; Koenighofer, M.; Vrabel, S.; Laccone, F.; Neesen, J.; Frei, K.; Roesch, S.; Dossena, S.; Schoefer, C.; Lucas, T.; et al. Mutational spectrum in patients with autosomal dominant hearing loss in Austria. Genes 2021. submitted. [Google Scholar]
- Doucette, L.; Merner, N.D.; Cooke, S.; Ives, E.; Galutira, D.; Walsh, V.; Walsh, T.; MacLaren, L.; Cater, T.; Fernandez, B.; et al. Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is caused by a novel missense mutation in the Usher syndrome type IF gene PCDH15. Eur. J. Hum. Genet. 2009, 17, 554–564. [Google Scholar] [CrossRef]
- Friedman, T.B.; Belyantseva, I.A.; Frolenkov, G.I. Myosins and Hearing. Adv. Exp. Med. Biol. 2020, 1239, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cortez, R.L.P.; Yarza, T.K.L.; Bootpetch, T.C.; Tantoco, M.L.C.; Mohlke, K.L.; Cruz, T.L.G.; Chiong Perez, M.E.; Chan, A.L.; Lee, N.R.; Tobias-Grasso, C.A.M.; et al. Identification of novel candidate genes and variants for hearing loss and temporal bone anomalies. Genes 2021, 12, 566. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roesch, S.; Rasp, G.; Sarikas, A.; Dossena, S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiol. Res. 2021, 11, 423-442. https://doi.org/10.3390/audiolres11030040
Roesch S, Rasp G, Sarikas A, Dossena S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiology Research. 2021; 11(3):423-442. https://doi.org/10.3390/audiolres11030040
Chicago/Turabian StyleRoesch, Sebastian, Gerd Rasp, Antonio Sarikas, and Silvia Dossena. 2021. "Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review" Audiology Research 11, no. 3: 423-442. https://doi.org/10.3390/audiolres11030040