
Clinical and Molecular Characteristics of Patients with PLN R14del Cardiomyopathy: State-of-the-Art Review

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology
3. Clinical Manifestations
3.1. Signs and Symptoms
3.2. Electrocardiography
3.3. Cardiac Magnetic Resonance
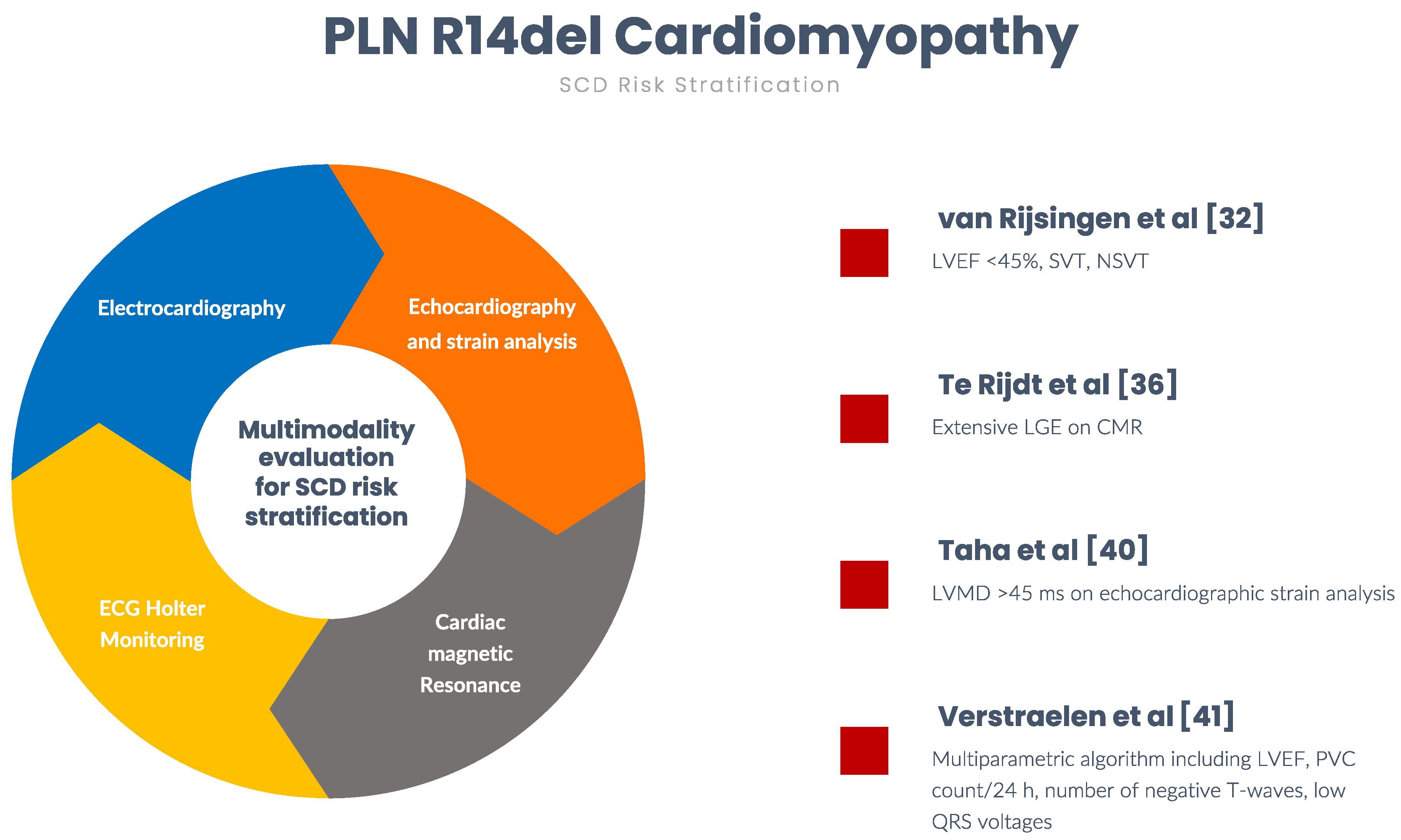
4. Risk Stratification for Sudden Cardiac Death
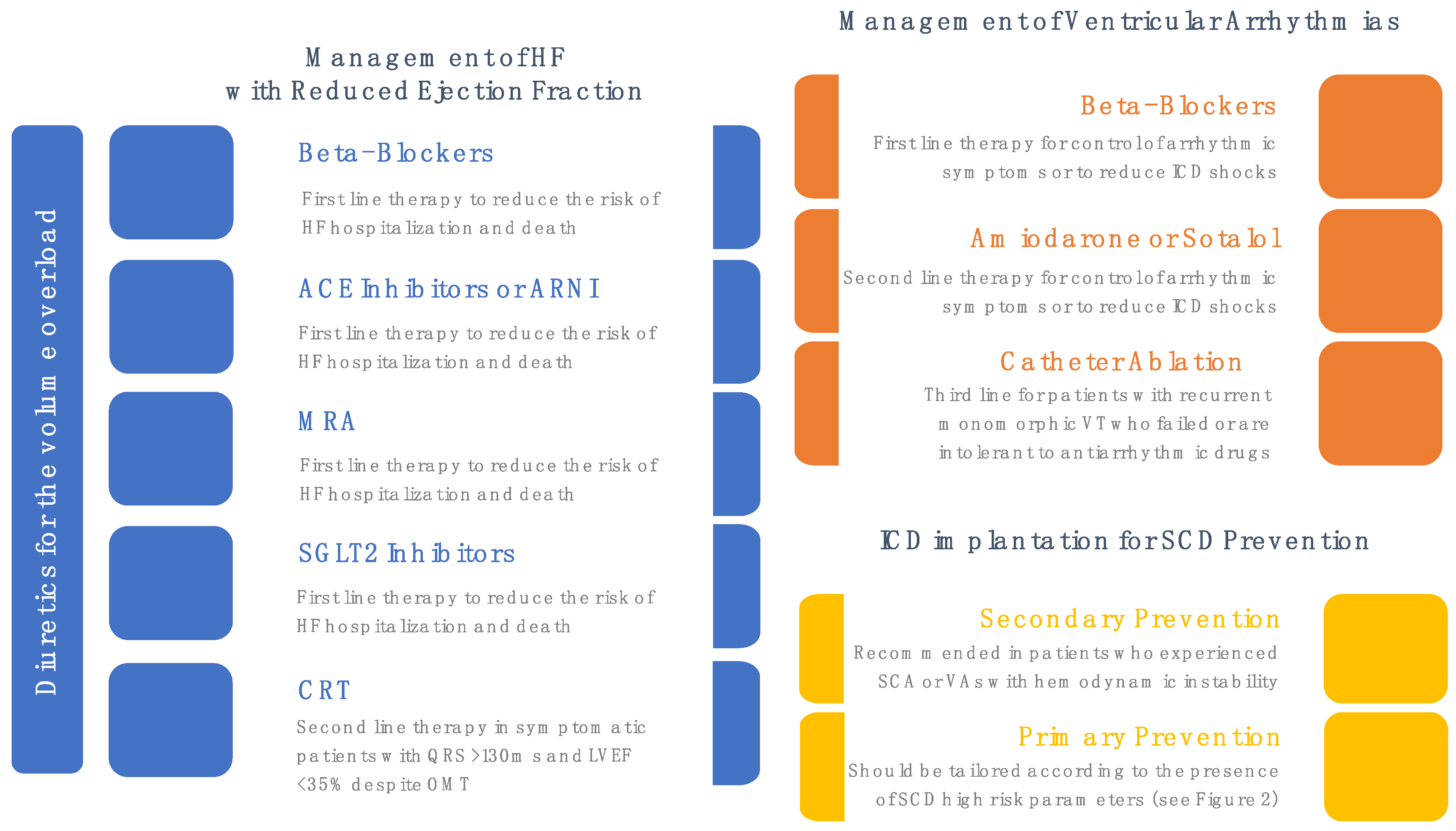
5. Medical Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef] [Green Version]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.; Ackerman, M.J.; Calkins, H.; Darrieux, F.; Daubert, J.P.; De Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.; Macrae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Genga, M.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Aranda, R.S.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. International Experts. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Zorzi, A.; Cipriani, A.; Bauce, B.; Bariani, R.; Beffagna, G.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; et al. Evolving Diagnostic Criteria for Arrhythmogenic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e021987. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.W.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Ast, J.F.H.-V.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Chen, S.N.; Ferro, M.D.; PaldinoMD, A.; De Angelis, G.; Brun, F.; et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.-C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Van Opbergen, C.J.M.; Delmar, M.; Van Veen, T.A.B. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: Focus on calcium sensitive pathways. Neth. Heart J. 2017, 25, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Eijgenraam, T.R.; Boukens, B.J.; Boogerd, C.J.; Schouten, E.M.; Van De Kolk, C.W.A.; Stege, N.M.; Rijdt, W.P.T.; Hoorntje, E.T.; Van Der Zwaag, P.A.; Van Rooij, E.; et al. The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Sci. Rep. 2020, 10, 9819. [Google Scholar] [CrossRef]
- Maier, L.S.; Bers, D. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyen, D.A.; Perea-Gil, I.; Maas, R.G.; Harakalova, M.; Gavidia, A.A.; Ataam, J.A.; Wu, T.-H.; Vink, A.; Pei, J.; Vadgama, N.; et al. Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy. Circulation 2021, 144, 382–392. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Eijgenraam, T.R.; Boogerd, C.J.; Stege, N.M.; Teixeira, V.O.N.; Dokter, M.M.; Schmidt, L.E.; Yin, X.; Theofilatos, K.; Mayr, M.; van der Meer, P.; et al. Protein Aggregation Is an Early Manifestation of Phospholamban p.(Arg14del)-Related Cardiomyopathy: Development of PLN-R14del-Related Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008532. [Google Scholar] [CrossRef]
- Cuello, F.; Knaust, A.E.; Saleem, U.; Loos, M.; Raabe, J.; Mosqueira, D.; Laufer, S.; Schweizer, M.; van der Kraak, P.; Flenner, F.; et al. Impairment of the ER/mitochondria compartment in human cardiomyocytes with PLN p.Arg14del mutation. EMBO Mol. Med. 2021, 13, e13074. [Google Scholar] [CrossRef]
- Van Der Zwaag, P.A.; Van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; Van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; Van Lochem, L.T.; De Boer, R.A.; Hofstra, R.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Rijdt, W.P.T.; Hoorntje, E.T.; de Brouwer, R.; Oomen, A.; Amin, A.; van der Heijden, J.F.; Karper, J.C.; Westenbrink, B.D.; Silljé, H.H.W.; Riele, A.S.J.M.T.; et al. Rationale and design of the PHOspholamban RElated CArdiomyopathy intervention STudy (i-PHORECAST). Neth. Heart J. 2021, 30, 84–95. [Google Scholar] [CrossRef]
- van Rijsingen, I.A.; van der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.; Zwinderman, A.H.; Pinto, Y.M.; Deprez, R.H.L.D.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers: Results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Sepehrkhouy, S.; Gho, J.M.; van Es, R.; Harakalova, M.; de Jonge, N.; Dooijes, D.; van der Smagt, J.J.; Buijsrogge, M.P.; Hauer, R.N.; Goldschmeding, R.; et al. Distinct fibrosis pattern in desmosomal and phospholamban mutation carriers in hereditary cardiomyopathies. Heart Rhythm 2017, 14, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Perrot, A.; Geier, C.; Boldt, L.-H.; Schmidt, G.; Lehmkuhl, H.B.; Hetzer, R.; Dietz, R.; Gutberlet, M.; Haverkamp, W.; et al. Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Heart Rhythm 2009, 6, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Rijdt, W.P.T.; Sande, J.N.T.; Gorter, T.M.; Van Der Zwaag, P.A.; Van Rijsingen, I.A.; Boekholdt, M.; Van Tintelen, J.P.; Van Haelst, P.L.; Planken, R.N.; De Boer, R.A.; et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: Phenotypic insights from cardiovascular magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 92–100. [Google Scholar] [CrossRef] [PubMed]
- van de Leur, R.R.; Taha, K.; Bos, M.N.; van der Heijden, J.F.; Gupta, D.; Cramer, M.J.; Hassink, R.J.; van der Harst, P.; Doevendans, P.A.; Asselbergs, F.W.; et al. Discovering and Visualizing Disease-Specific Electrocardiogram Features Using Deep Learning: Proof-of-Concept in Phospholamban Gene Mutation Carriers. Circ. Arrhythmia Electrophysiol. 2021, 14, e009056. [Google Scholar] [CrossRef] [PubMed]
- Bourfiss, M.; Riele, A.S.T.; Mast, T.P.; Cramer, M.J.; Van Der Heijden, J.F.; Van Veen, T.A.; Loh, P.; Dooijes, D.; Hauer, R.N.; Velthuis, B.K. Influence of Genotype on Structural Atrial Abnormalities and Atrial Fibrillation or Flutter in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2016, 27, 1420–1428. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef]
- Taha, K.; Verstraelen, T.E.; de Brouwer, R.; de Bruin-Bon, R.H.A.C.M.; Cramer, M.J.; Te Rijdt, W.P.; Bouma, B.J.; de Boer, R.A.; Doevendans, P.A.; Asselbergs, F.W.; et al. Optimal echocardiographic assessment of myocardial dysfunction for arrhythmic risk stratification in phospholamban mutation carriers. Eur. Heart J. Cardiovasc. Imaging 2021. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, T.E.; van Lint, F.H.M.; Bosman, L.P.; de Brouwer, R.; Proost, V.M.; Abeln, B.G.S.; Taha, K.; Zwinderman, A.H.; Dickhoff, C.; Oomen, T.; et al. Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriers-reaching the frontiers of individual risk prediction. Eur. Heart J. 2021, 42, 2842–2850. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, E.K.; Wozniak, O.; Konka, M.; Rydlewska-Sadowska, W.; Biederman, A.; Hoffman, P. Thromboembolic complications in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Europace 2006, 8, 596–600. [Google Scholar] [CrossRef]
- Moss, A.J.; Schuger, C.; Beck, C.A.; Brown, M.W.; Cannom, D.S.; Daubert, J.P.; Estes, N.M.; Greenberg, H.; Hall, W.J.; Huang, D.T.; et al. Reduction in inappropriate therapy and mortality through ICD programming. N. Engl. J. Med. 2012, 367, 2275–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparini, M.; Proclemer, A.; Klersy, C.; Kloppe, A.; Lunati, M.; Ferrer, J.B.M.; Hersi, A.; Gulaj, M.; Wijfels, M.C.E.F.; Santi, E.; et al. Effect of long-detection interval vs standard-detection interval for implantable cardioverter-defibrillators on antitachycardia pacing and shock delivery: The ADVANCE III randomized clinical trial. JAMA 2013, 309, 1903–1911. [Google Scholar] [CrossRef]
- Ruwald, M.H.; Abu-Zeitone, A.; Jons, C.; Ruwald, A.-C.; McNitt, S.; Kutyifa, V.; Zareba, W.; Moss, A.J. Impact of carvedilol and metoprolol on inappropriate implantable cardioverter-defibrillator therapy: The MADIT-CRT trial (Multicenter Automatic Defibrillator Implantation with Cardiac Resynchronization Therapy). J. Am. Coll. Cardiol. 2013, 62, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, G.M.; Glidden, D.; Polonsky, B.; Zareba, W.; Smith, L.M.; Cannom, D.S.; Estes, N.M., III; Marcus, F.; Scheinman, M.M. Multidisciplinary Study of Right Ventricular Dysplasia Investigators. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: A report from the North American ARVC Registry. J. Am. Coll. Cardiol. 2009, 54, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.J.; Dorian, P.; Roberts, R.S.; Gent, M.; Bailin, S.; Fain, E.S.; Thorpe, K.; Champagne, J.; Talajic, M.; Coutu, B.; et al. Optimal Pharmacological Therapy in Cardioverter Defibrillator Patients (OPTIC) Investigators. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study: A randomized trial. JAMA 2006, 295, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Tung, R.; Vaseghi, M.; Frankel, D.S.; Vergara, P.; Di Biase, L.; Nagashima, K.; Yu, R.; Vangala, S.; Tseng, C.-H.; Choi, E.-K.; et al. Freedom from recurrent ventricular tachycardia after catheter ablation is associated with improved survival in patients with structural heart disease: An International VT Ablation Center Collaborative Group study. Heart Rhythm 2015, 12, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monda, E.; Blasi, E.; De Pasquale, A.; Di Vilio, A.; Amodio, F.; Caiazza, M.; Diana, G.; Lioncino, M.; Perna, A.; Verrillo, F.; et al. Clinical and Molecular Characteristics of Patients with PLN R14del Cardiomyopathy: State-of-the-Art Review. Cardiogenetics 2022, 12, 112-121. https://doi.org/10.3390/cardiogenetics12010012
Monda E, Blasi E, De Pasquale A, Di Vilio A, Amodio F, Caiazza M, Diana G, Lioncino M, Perna A, Verrillo F, et al. Clinical and Molecular Characteristics of Patients with PLN R14del Cardiomyopathy: State-of-the-Art Review. Cardiogenetics. 2022; 12(1):112-121. https://doi.org/10.3390/cardiogenetics12010012
Chicago/Turabian StyleMonda, Emanuele, Ettore Blasi, Antonio De Pasquale, Alessandro Di Vilio, Federica Amodio, Martina Caiazza, Gaetano Diana, Michele Lioncino, Alessia Perna, Federica Verrillo, and et al. 2022. "Clinical and Molecular Characteristics of Patients with PLN R14del Cardiomyopathy: State-of-the-Art Review" Cardiogenetics 12, no. 1: 112-121. https://doi.org/10.3390/cardiogenetics12010012