


Lipid Nanocarriers-Enabled Delivery of Antibiotics and Antimicrobial Adjuvants to Overcome Bacterial Biofilms
 , , , and
, , , and
Abstract
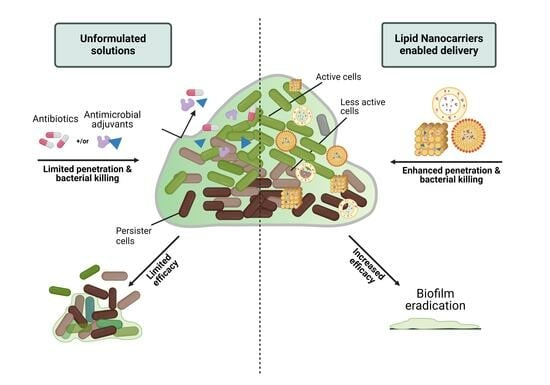
:
1. Introduction
2. Biofilms—Definition, Composition, Life Cycle, and Therapeutic Challenges
3. Various Approaches to Combat Biofilms
3.1. Antimicrobial Adjuvants to Improve Biofilm Killing
3.1.1. Quorum Sensing Inhibitors
3.1.2. EPS-Degrading Enzymes
Glycosidases
- Dispersion B (DspB)
- PgaB
- Alginate lyase (AL)
- Ps1G and Pe1A
Deoxyribonucleases
Proteases
4. Lipid Nanocarriers Mediated Delivery of Antibiotics and Antimicrobial Adjuvants
4.1. Liposomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanocarrier | Encapsulated Agent | Class | Biofilm | Testing Method | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Liposomes | Ps1G + Tobramycin | Biofilm-dispersing enzyme + Aminoglycoside antibiotic | P. aeruginosa | CV assay, MBEC assay | Improved the activity of tobramycin with a 20% reduction in the biofilm biomass. | [13] |
| Liposomes | Lysozyme + Gentamicin | Biofilm-dispersing enzyme + Aminoglycoside antibiotic | P. aeruginosa and S. aureus | CV assay, MTT assay | Liposomal formulation significantly reduced the biofilm biomass and live bacterial count compared to free drugs and enzymes. | [190] |
| Liposome | N-Acetylcysteine + Tobramycin | Antibiotic adjuvant + Aminoglycoside antibiotic | Escherichia coli, Acinetobacter baumannii, and Klebsiella pneumoniae | CV assay | Tobramycin and encapsulated liposomes significantly reduced the biofilm biomass and showed enhanced efficacy in inhibiting biofilm formation compared to unformulated drugs. | [195] |
| Liposome | N-Acetylcysteine + Azithromycin | Antibiotic adjuvant + antibiotic | E. coli (Clinical isolate) | CV assay | Azithromycin encapsulating in liposomes demonstrated higher biofilm reduction i.e., 93.22%) at 1X MIC. | [196] |
| liposome | Lysozyme + Chlorhexidine + Lactoferrin | EPS-degrading enzyme + antibiotic + glycoprotein | Streptococcus mutans, Streptococcus sobrinus | CFU enumeration | Encapsulated chlorhexidine completely inhibited the biofilm formation. | [197] |
| Liposome | Serratiopeptidase (SRP) + Levofloxacin | EPS-degrading enzyme + antibiotic | S. aureus infected rats | CV assay | Levofloxacin (sub-MIC concentration) co-encapsulated with SPR significantly eradicated the preformed biofilm i.e., >90%. | [198] |
| Liposome | DNase I and proteinase K | EPS-degrading enzymes | Cutibacterium acnes | CV assay, Porcine skin model (in-vitro), Murine skin and catheters (In vivo) | Dual enzyme-loaded liposomes exhibited greater biofilm-formation inhibition and deeper penetration (85%) into biofilm thickness. Enhanced penetration and facile delivery into porcine skin in-vitro. Potent in-vivo activity in eliminating colonization of C. acnes in murine skin and catheters with a 2-log reduction in CFU in catheters treated with liposomes compared to untreated controls. | [193] |
| Liposome | CDC and PF | Quorum sensing inhibitors | P. aeruginosa | CV assay | Liposomal formulations demonstrated dose-dependent anti-biofilm activity compared to fee QS inhibitors. | [108] |
| Liposome | 2-nitroimidazole derivative, 6-NIH + DETA NONOate + Azithromycin | Antibiotic adjuvant + biofilm dispersant + antibiotic | P. aeruginosa | CV assay, CLSM | Liposomal formulation significantly eradicated mature biofilm, efficiently killed dispersed bacteria, inhibited the metabolism of survivors, and also inhibited recurrent infection by preventing bacteria from adhering to airway epithelial cells. | [199] |
| Liposome | Farnesol + Ciprofloxacin | QSI + Quinolone antibiotic | P. aeruginosa | XTT reduction assay, Confocal laser scanning microscopy (CLSM) | 80% reduction of P. aeruginosa biofilm biomass at 0.128 µg/mL concentration of ciprofloxacin. Greater cell death was observed via CLSM imaging after the biofilm treatment with the formulation compared to the liposomal ciprofloxacin alone. | [194] |
| Liposome | Bismuth ethanedithiol (BiEDT) + Tobramycin | Antimicrobial adjuvant + antibiotic | P. aeruginosa | QS and Virulence factor assay (In vitro), Sprague Dawley rats (In vivo) | Encapsulated liposomes effectively disrupt the quorum sensing and significantly reduce the chitinase, lipase, and protease production (In-vitro) with a 3-log CFU reduction (In-vivo) compared to unformulated drugs. | [200] |
| Liposome | Bismuth-ethanedithiol + Alginate lyase + Tobramycin | Antimicrobial adjuvant + EPS-degrading enzyme + antibiotic | Mucoid P. aeruginosa (Clinical isolate) | MBEC assay, Carbazole assay | The anti-biofilm activity of bismuth-ethanedithiol + TOB compared with unformulated TOB was decreased by 4–32-fold. While addition of enzymes markedly increased the biofilm eradication. | [201] |
| LCNPs | Alginate lyase + Gentamicin | Biofilm-dispersing enzyme + Aminoglycoside antibiotic | Mucoid P. aeruginosa (Clinical isolate) | CV assay, MBEC assay | Infection-responsive antibiotic release, >2-log decline in P. aeruginosa biofilm compared to unformulated solutions. | [202] |
| LCNPs | Ps1G + Tobramycin | Biofilm-dispersing enzyme + Aminoglycoside antibiotic | P. aeruginosa (PAO1 and PAO1 PBADpsl ΔpelF | CV assay, MBEC assay (In-vitro) C. elegans (In-vivo) | Enhanced the antimicrobial efficacy of tobramycin by 10–100 folds and enhanced the P. aeruginosa-infected C. elegans survival. | [13] |
| Squalenyl Hydrogen Sulfate Nanoparticles | Alkylquinolone QS+ Tobramycin | QSI + Aminoglycoside antibiotic | P. aeruginosa | MBEC assay | Three-fold higher penetration and completely eradicated P. aeruginosa biofilms at almost eight times lower concentrations of tobramycin than the free drug and QSI alone | [203] |
| SLNPs | DNase I and levofloxacin | Enzyme + antibiotic | P. aeruginosa and S. aureus | Alamar-blue assay | SLNP formulation significantly increased the levofloxacin efficacy and markedly reduced the S. aureus and P. aeruginosa biofilm formation. | [204] |
| SLNPs | cis-2-decanoic acid (C2DA) + rifampicin | QSI inhibitor + antibiotic | S. aureus and S. epidermidis | CV assay | Demonstrated better anti-biofilm activity than free agents particularly in the biofilm formation stage, while unable to remove the preformed biofilms. | [205] |
| SLNPs | Anacardic acid + DNase | Antimicrobial agent + EPS-degrading enzyme | S. aureus | MBEC assay | Significantly reduced the MBIC and MBEC and markedly reduced the biofilm thickness and biomass demonstrated by CLSM | [206] |
| NLCs | DNase I and levofloxacin | Enzyme + antibiotic | P. aeruginosa | Alamar-blue assay | This formulation exhibited improved anti-biofilm activity against cystic fibrosis by decreasing the viscoelasticity in the patient’s lungs. | [204] |
| LNPs | N-acetyl-l-cysteine (NAC) + Moxifloxacin | Antimicrobial adjuvant + antibiotic | S. epidermidis, and P. aeruginosa | MBEC assay, CV assay, SEM biofilm analysis, MTT assay | NAC-loaded and unloaded moxifloxacin-LNPs significantly reduced the viable bacterial count, with no significant difference between the two. But NAC-loaded LNPs exhibited a safer profile compared to unloaded LNPs which is promising for in-vivo application. | [207] |
| Nanoemulsion | Eucalyptus globulus oil | Anti-biofilm agent | P. aeruginosa and Candida spp. | Calcofluor staining, atomic force microscopy. | Nanoencpsulated oil demonstrated improved anti-biofilm activity against Candida spp. (10-fold reduction in CFU) but was infective against P. aeruginosa biofilm due to less oil concentration (only 5%) being ineffective. | [208] |
| Nanosphere | Acylase + Gentamicin | QQ enzyme + Aminoglycoside antibiotics | P. aeruginosa | CV assay, fluorescent microscopy | Inhibit 95% of P. aeruginosa biofilm biomass production at 0.125 × 1013 NSs mL−1. | [209] |
4.2. Lyotropic Liquid Crystal Nanoparticles
4.3. Solid-Lipid Nanoparticles
4.4. Nanostructured Lipid Carriers
4.5. Other Novel LNCs
5. Current Perspective and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Virulence mechanisms of bacterial pathogens. Microbiol. Spectr. 2016, 2016, 481–511. [Google Scholar]
- Tornimbene, B.; Eremin, S.; Escher, M.; Griskeviciene, J.; Manglani, S.; Pessoa-Silva, C.L. WHO global antimicrobial resistance surveillance system early implementation 2016–17. Lancet Infect. Dis. 2018, 18, 241–242. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Organisation for Economic Co-operation and Development; Staff, D. Stemming the Superbug Tide: Just a Few Dollars More; OECD: Paris, France, 2019. [Google Scholar]
- Lampi, E.; Carelli, D.; Pierre, J.; Rönnerstrand, B. Two pandemics: The COVID-19 pandemic’s impact on future AMR collaboration in Europe. Humanit. Soc. Sci. Commun. 2023, 10, 441. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Birkelbach, J.; Walesch, S.; Müller, R. Current developments in antibiotic discovery: Global microbial diversity as a source for evolutionary optimized anti-bacterial. EMBO Rep. 2023, 24, e56184. [Google Scholar] [CrossRef]
- Choi, V.; Rohn, J.L.; Stoodley, P.; Carugo, D.; Stride, E. Drug delivery strategies for antibiofilm therapy. Nat. Rev. Microbiol. 2023, 21, 555–572. [Google Scholar] [CrossRef]
- Ciofu, O.; Moser, C.; Jensen, P.Ø.; Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 2022, 20, 621–635. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef]
- Nourbakhsh, F.; Nasrollahzadeh, M.S.; Tajani, A.S.; Soheili, V.; Hadizadeh, F. Bacterial biofilms and their resistance mechanisms: A brief look at treatment with natural agents. Folia Microbiol. 2022, 67, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. Pharm. Ther. 2015, 40, 344. [Google Scholar]
- Mah, T.-F.C.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Thorn, C.R.; Raju, D.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Howell, P.L.; Prestidge, C.A.; Thomas, N. Protective liquid crystal nanoparticles for targeted delivery of PslG: A biofilm dispersing enzyme. ACS Infect. Dis. 2021, 7, 2102–2115. [Google Scholar] [CrossRef]
- Sauer, K.; Stoodley, P.; Goeres, D.M.; Hall-Stoodley, L.; Burmølle, M.; Stewart, P.S.; Bjarnsholt, T. The biofilm life cycle: Expanding the conceptual model of biofilm formation. Nat. Rev. Microbiol. 2022, 20, 608–620. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Moser, C.; Bassi, G.; Coenye, T.; Donelli, G.; Hall-Stoodley, L.; Holá, V.; Imbert, C.; Kirketerp-Møller, K. ESCMID guideline for the diagnosis and treatment of biofilm infections 2014. Clin. Microbiol. Infect. 2015, 21, S1–S25. [Google Scholar] [CrossRef]
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Current update on intrinsic and acquired colistin resistance mechanisms in bacteria. Front. Med. 2021, 8, 677720. [Google Scholar] [CrossRef]
- Shariati, A.; Dadashi, M.; Moghadam, M.T.; van Belkum, A.; Yaslianifard, S.; Darban-Sarokhalil, D. Global prevalence and distribution of vancomycin resistant, vancomycin intermediate and heterogeneously vancomycin intermediate Staphylococcus aureus clinical isolates: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12689. [Google Scholar] [CrossRef]
- Thu, P.N.T.; Huong, M.N.T.; Thi, N.T.; Thanh, H.N.; Minh, K.P. Combination antibiotic therapy versus monotherapy in the treatment of acute exacerbations of chronic obstructive pulmonary disease: An open-label randomized trial. BMC Infect. Dis. 2021, 21, 1019. [Google Scholar] [CrossRef]
- Tyers, M.; Wright, G.D. Drug combinations: A strategy to extend the life of antibiotics in the 21st century. Nat. Rev. Microbiol. 2019, 17, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, G.; Acharya, Y.; Haldar, J. Antibiotic Adjuvants: A Versatile Approach to Combat Antibiotic Resistance. ACS Omega 2023, 8, 10757–10783. [Google Scholar] [CrossRef]
- Ferreira, I.S.; Bettencourt, A.; Bétrisey, B.; Gonçalves, L.M.; Trampuz, A.; Almeida, A.J. Improvement of the antibacterial activity of daptomycin-loaded polymeric microparticles by Eudragit RL 100: An assessment by isothermal microcalorimetry. Int. J. Pharm. 2015, 485, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Natan, M.; Banin, E. From Nano to Micro: Using nanotechnology to combat microorganisms and their multidrug resistance. FEMS Microbiol. Rev. 2017, 41, 302–322. [Google Scholar] [CrossRef]
- Xiong, M.-H.; Bao, Y.; Yang, X.-Z.; Zhu, Y.-H.; Wang, J. Delivery of antibiotics with polymeric particles. Adv. Drug Deliv. Rev. 2014, 78, 63–76. [Google Scholar] [CrossRef]
- Malaekeh-Nikouei, B.; Bazzaz, B.S.F.; Mirhadi, E.; Tajani, A.S.; Khameneh, B. The role of nanotechnology in combating biofilm-based antibiotic resistance. J. Drug Deliv. Sci. Technol. 2020, 60, 101880. [Google Scholar] [CrossRef]
- Makabenta, J.M.V.; Nabawy, A.; Li, C.-H.; Schmidt-Malan, S.; Patel, R.; Rotello, V.M. Nanomaterial-based therapeutics for antibiotic-resistant bacterial infections. Nat. Rev. Microbiol. 2021, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-Y.; Van der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-based antimicrobial delivery-systems for the treatment of bacterial infections. Front. Chem. 2020, 7, 872. [Google Scholar] [CrossRef]
- Kirtane, A.R.; Verma, M.; Karandikar, P.; Furin, J.; Langer, R.; Traverso, G. Nanotechnology approaches for global infectious diseases. Nat. Nanotechnol. 2021, 16, 369–384. [Google Scholar] [CrossRef]
- Thorn, C.R.; Thomas, N.; Boyd, B.J.; Prestidge, C.A. Nano-fats for bugs: The benefits of lipid nanoparticles for antimicrobial therapy. Drug Deliv. Transl. Res. 2021, 11, 1598–1624. [Google Scholar] [CrossRef] [PubMed]
- Arana, L.; Gallego, L.; Alkorta, I. Incorporation of antibiotics into solid lipid nanoparticles: A promising approach to reduce antibiotic resistance emergence. Nanomaterials 2021, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.V.; Gunawan, C.; Mann, R. We are one: Multispecies metabolism of a biofilm consortium and their treatment strategies. Front. Microbiol. 2021, 12, 635432. [Google Scholar] [CrossRef]
- Forier, K.; Raemdonck, K.; De Smedt, S.C.; Demeester, J.; Coenye, T.; Braeckmans, K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J. Control. Release 2014, 190, 607–623. [Google Scholar] [CrossRef]
- Fulaz, S.; Vitale, S.; Quinn, L.; Casey, E. Nanoparticle–biofilm interactions: The role of the EPS matrix. Trends Microbiol. 2019, 27, 915–926. [Google Scholar] [CrossRef]
- Huang, R.; Li, M.; Gregory, R.L. Bacterial interactions in dental biofilm. Virulence 2011, 2, 435–444. [Google Scholar] [CrossRef]
- Dwivedi, D.; Sehgal, T. Biofilm Development in Gram-Positive and Gram-Negative Bacteria. In Focus on Bacterial Biofilms; IntechOpen: London, UK, 2022. [Google Scholar]
- Abdalla, A.K.; Ayyash, M.M.; Olaimat, A.N.; Osaili, T.M.; Al-Nabulsi, A.A.; Shah, N.P.; Holley, R. Exopolysaccharides as antimicrobial agents: Mechanism and spectrum of activity. Front. Microbiol. 2021, 12, 664395. [Google Scholar] [CrossRef]
- Singh, S.; Singh, S.K.; Chowdhury, I.; Singh, R. Understanding the mechanism of bacterial biofilms resistance to antimicrobial agents. Open Microbiol. J. 2017, 11, 53. [Google Scholar] [CrossRef]
- Quan, K.; Hou, J.; Zhang, Z.; Ren, Y.; Peterson, B.W.; Flemming, H.-C.; Mayer, C.; Busscher, H.J.; van der Mei, H.C. Water in bacterial biofilms: Pores and channels, storage and transport functions. Crit. Rev. Microbiol. 2022, 48, 283–302. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The microbial “protective clothing” in extreme environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef] [PubMed]
- Crabbé, A.; Jensen, P.Ø.; Bjarnsholt, T.; Coenye, T. Antimicrobial tolerance and metabolic adaptations in microbial biofilms. Trends Microbiol. 2019, 27, 850–863. [Google Scholar] [CrossRef]
- Shree, P.; Singh, C.K.; Sodhi, K.K.; Surya, J.N.; Singh, D.K. Biofilms: Understanding the structure and contribution towards bacterial resistance in antibiotics. Med. Microecol. 2023, 16, 100084. [Google Scholar] [CrossRef]
- Stewart, P.S. A review of experimental measurements of effective diffusive permeabilities and effective diffusion coefficients in biofilms. Biotechnol. Bioeng. 1998, 59, 261–272. [Google Scholar] [CrossRef]
- Li, P.; Yin, R.; Cheng, J.; Lin, J. Bacterial Biofilm Formation on Biomaterials and Approaches to Its Treatment and Prevention. Int. J. Mol. Sci. 2023, 24, 11680. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M. Role of biofilms in antimicrobial resistance. ASAIO J. 2000, 46, S47–S52. [Google Scholar] [CrossRef] [PubMed]
- Espuelas, M.; Legrand, P.; Loiseau, P.; Bories, C.; Barratt, G.; Irache, J. In vitro antileishmanial activity of amphotericin B loaded in poly (ε-caprolactone) nanospheres. J. Drug Target. 2002, 10, 593–599. [Google Scholar] [CrossRef]
- Hunter, R.C.; Klepac-Ceraj, V.; Lorenzi, M.M.; Grotzinger, H.; Martin, T.R.; Newman, D.K. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. Am. J. Respir. Cell Mol. Biol. 2012, 47, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Deepigaa, M. Antibacterial resistance of bacteria in biofilms. Res. J. Pharm. Technol. 2017, 10, 4019–4023. [Google Scholar] [CrossRef]
- Tresse, O.; Jouenne, T.; Junter, G.-A. The role of oxygen limitation in the resistance of agar-entrapped, sessile-like Escherichia coli to aminoglycoside and β-lactam antibiotics. J. Antimicrob. Chemother. 1995, 36, 521–526. [Google Scholar] [CrossRef]
- Pandey, R.; Khuller, G. Solid lipid particle-based inhalable sustained drug delivery system against experimental tuberculosis. Tuberculosis 2005, 85, 227–234. [Google Scholar] [CrossRef]
- Reid, G. Biofilms in infectious disease and on medical devices. Int. J. Antimicrob. Agents 1999, 11, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar]
- Haque, S.; Yadav, D.K.; Bisht, S.C.; Yadav, N.; Singh, V.; Dubey, K.K.; Jawed, A.; Wahid, M.; Dar, S.A. Quorum sensing pathways in Gram-positive and-negative bacteria: Potential of their interruption in abating drug resistance. J. Chemother. 2019, 31, 161–187. [Google Scholar] [CrossRef]
- Lami, R.; Urios, L.; Molmeret, M.; Grimaud, R. Quorum sensing in biofilms: A key mechanism to target in ecotoxicological studies. Crit. Rev. Microbiol. 2023, 49, 786–804. [Google Scholar] [CrossRef]
- Wang, Y. Liposome as a delivery system for the treatment of biofilm-mediated infections. J. Appl. Microbiol. 2021, 131, 2626–2639. [Google Scholar] [CrossRef]
- Lin, Y.-K.; Yang, S.-C.; Hsu, C.-Y.; Sung, J.-T.; Fang, J.-Y. The antibiofilm nanosystems for improved infection inhibition of microbes in skin. Molecules 2021, 26, 6392. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.; Fan, H.-M.; Tao, H.-R.; Huang, J.-D. Recent strategies to combat biofilms using antimicrobial agents and therapeutic approaches. Pathogens 2022, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.G.; Yousef, A.E. Combating Bacterial Biofilms: Current and Emerging Antibiofilm Strategies for Treating Persistent Infections. Antibiotics 2023, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Jawetz, E.; Gunnison, J.; Bruff, J.; Coleman, V. Studies on antibiotic synergism and antagonism: Synergism among seven antibiotics against various bacteria in vitro. J. Bacteriol. 1952, 64, 29–39. [Google Scholar] [CrossRef]
- Bushby, S.; Hitchings, G. Trimethoprim, a sulphonamide potentiator. Br. J. Pharmacol. Chemother. 1968, 33, 72. [Google Scholar] [CrossRef]
- Kerantzas, C.A.; Jacobs, W.R., Jr. Origins of combination therapy for tuberculosis: Lessons for future antimicrobial development and application. MBio 2017, 8, e01586-16. [Google Scholar] [CrossRef]
- Kumar, V.; Yasmeen, N.; Pandey, A.; Chaudhary, A.A.; Alawam, A.S.; Rudayni, H.A.; Islam, A.; Lakhawat, S.S.; Sharma, P.K.; Shahid, M. Antibiotic adjuvants: Synergistic tool to combat multi-drug resistant pathogens. Front. Cell. Infect. Microbiol. 2023, 13, 1293633. [Google Scholar] [CrossRef]
- Singh, N.; Romero, M.; Travanut, A.; Monteiro, P.F.; Jordana-Lluch, E.; Hardie, K.R.; Williams, P.; Alexander, M.R.; Alexander, C. Dual bioresponsive antibiotic and quorum sensing inhibitor combination nanoparticles for treatment of Pseudomonas aeruginosa biofilms in vitro and ex vivo. Biomater. Sci. 2019, 7, 4099–4111. [Google Scholar] [CrossRef]
- De la Fuente-Núñez, C.S.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef]
- Reffuveille, F.; De La Fuente-Nú̃nez, C.; Mansour, S.; Hancock, R.E. A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob. Agents Chemother. 2014, 58, 5363–5371. [Google Scholar] [CrossRef]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic resistance breakers: Current approaches and future directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief overview of approaches and challenges in new antibiotic development: A focus on drug repurposing. Front. Cell. Infect. Microbiol. 2021, 11, 684515. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Bose, S.; Shaoo, A.; Das, S.K. Nanotechnology based therapeutic approaches: An advanced strategy to target the biofilm of ESKAPE pathogens. Mater. Adv. 2023, 4, 2544–2572. [Google Scholar] [CrossRef]
- Ruhal, R.; Kataria, R. Biofilm patterns in gram-positive and gram-negative bacteria. Microbiol. Res. 2021, 251, 126829. [Google Scholar] [CrossRef]
- Giaouris, E.E.; Simões, M.V. Pathogenic biofilm formation in the food industry and alternative control strategies. In Foodborne Diseases; Elsevier: Amsterdam, The Netherlands, 2018; pp. 309–377. [Google Scholar]
- LaSarre, B.; Federle, M.J. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 2013, 77, 73–111. [Google Scholar] [CrossRef] [PubMed]
- Başaran, T.I.; Berber, D.; Gökalsın, B.; Tramice, A.; Tommonaro, G.; Abbamondi, G.R.; Erginer Hasköylü, M.; Toksoy Öner, E.; Iodice, C.; Sesal, N.C. Extremophilic Natrinema versiforme against Pseudomonas aeruginosa quorum sensing and biofilm. Front. Microbiol. 2020, 11, 79. [Google Scholar] [CrossRef]
- Cáceres, M.; Hidalgo, W.; Stashenko, E.; Torres, R.; Ortiz, C. Essential oils of aromatic plants with antibacterial, anti-biofilm and anti-quorum sensing activities against pathogenic bacteria. Antibiotics 2020, 9, 147. [Google Scholar] [CrossRef]
- Zhong, L.; Ravichandran, V.; Zhang, N.; Wang, H.; Bian, X.; Zhang, Y.; Li, A. Attenuation of Pseudomonas aeruginosa quorum sensing by natural products: Virtual screening, evaluation and biomolecular interactions. Int. J. Mol. Sci. 2020, 21, 2190. [Google Scholar] [CrossRef]
- Kalia, V.C. Quorum sensing inhibitors: An overview. Biotechnol. Adv. 2013, 31, 224–245. [Google Scholar] [CrossRef] [PubMed]
- Scoffone, V.C.; Trespidi, G.; Chiarelli, L.R.; Barbieri, G.; Buroni, S. Quorum sensing as antivirulence target in cystic fibrosis pathogens. Int. J. Mol. Sci. 2019, 20, 1838. [Google Scholar] [CrossRef]
- Jakobsen, T.H.; Warming, A.N.; Vejborg, R.M.; Moscoso, J.A.; Stegger, M.; Lorenzen, F.; Rybtke, M.; Andersen, J.B.; Petersen, R.; Andersen, P.S. A broad range quorum sensing inhibitor working through sRNA inhibition. Sci. Rep. 2017, 7, 9857. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Zhang, P.; Bhagirath, A.Y.; Duan, K. ClpV3 of the H3-type VI secretion system (H3-T6SS) affects multiple virulence factors in Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 1096. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, A.; Moretta, A.; Canè, C.; Cirillo, A.; Duilio, A. Antimicrobial and antibiofilm peptides. Biomolecules 2020, 10, 652. [Google Scholar] [CrossRef]
- Yang, Y.-B.; Wang, S.; Wang, C.; Huang, Q.-Y.; Bai, J.-W.; Chen, J.-Q.; Chen, X.-Y.; Li, Y.-H. Emodin affects biofilm formation and expression of virulence factors in Streptococcus suis ATCC700794. Arch. Microbiol. 2015, 197, 1173–1180. [Google Scholar] [CrossRef]
- Yan, X.; Gu, S.; Shi, Y.; Cui, X.; Wen, S.; Ge, J. The effect of emodin on Staphylococcus aureus strains in planktonic form and biofilm formation in vitro. Arch. Microbiol. 2017, 199, 1267–1275. [Google Scholar] [CrossRef]
- Vasilchenko, A.S.; Rogozhin, E.A. Sub-inhibitory effects of antimicrobial peptides. Front. Microbiol. 2019, 10, 1160. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: Current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Shastry, R.P.; Rekha, P.; Rai, V.R. Biofilm inhibitory activity of metallo-protein AHL-lactonase from cell-free lysate of endophytic Enterobacter species isolated from Coscinium fenestratum Gaertn. Biocatal. Agric. Biotechnol. 2019, 18, 101009. [Google Scholar] [CrossRef]
- Ivanova, K.; Fernandes, M.M.; Francesko, A.; Mendoza, E.; Guezguez, J.; Burnet, M.; Tzanov, T. Quorum-quenching and matrix-degrading enzymes in multilayer coatings synergistically prevent bacterial biofilm formation on urinary catheters. ACS Appl. Mater. Interfaces 2015, 7, 27066–27077. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Zhao, A.; Wang, A.; Brown, Z.Z.; Muir, T.W.; Stone, H.A.; Bassler, B.L. Surface-attached molecules control Staphylococcus aureus quorum sensing and biofilm development. Nat. Microbiol. 2017, 2, 17080. [Google Scholar] [CrossRef] [PubMed]
- Krzyżek, P. Challenges and limitations of anti-quorum sensing therapies. Front. Microbiol. 2019, 10, 2473. [Google Scholar] [CrossRef]
- Imhann, F.; Vila, A.V.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; Van Dullemen, H.M. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Ismail, A.S.; Valastyan, J.S.; Bassler, B.L. A host-produced autoinducer-2 mimic activates bacterial quorum sensing. Cell Host Microbe 2016, 19, 470–480. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, P.; Nan, W.; Zhi, D.; Liu, R.; Li, H. The use of (5Z)-4-bromo-5-(bromomethylene)-2 (5H)-furanone for controlling acid mine drainage through the inhibition of Acidithiobacillus ferrooxidans biofilm formation. Bioresour. Technol. 2015, 186, 52–57. [Google Scholar] [CrossRef]
- Ali, I.A.; Cheung, B.P.; Matinlinna, J.; Lévesque, C.M.; Neelakantan, P. Trans-cinnamaldehyde potently kills Enterococcus faecalis biofilm cells and prevents biofilm recovery. Microb. Pathog. 2020, 149, 104482. [Google Scholar] [CrossRef]
- Mu, R.; Zhang, H.; Zhang, Z.; Li, X.; Ji, J.; Wang, X.; Gu, Y.; Qin, X. Trans-cinnamaldehyde loaded chitosan based nanocapsules display antibacterial and antibiofilm effects against cavity-causing Streptococcus mutans. J. Oral Microbiol. 2023, 15, 2243067. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Jayaprakasha, G.; Uckoo, R.M.; Patil, B.S. Inhibition of Escherichia coli O157: H7 motility and biofilm by β-sitosterol glucoside. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 5219–5228. [Google Scholar] [CrossRef]
- Ouyang, J.; Sun, F.; Feng, W.; Sun, Y.; Qiu, X.; Xiong, L.; Liu, Y.; Chen, Y. Quercetin is an effective inhibitor of quorum sensing, biofilm formation and virulence factors in Pseudomonas aeruginosa. J. Appl. Microbiol. 2016, 120, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.-B.; Shi, M.-Y.; Wang, W.-W.; Wu, L.-Y.; Bai, Y.-T.; Li, B.; Zhou, X.-Z.; Zhang, J.-Y. Novel quorum sensing inhibitor Echinatin as an antibacterial synergist against Escherichia coli. Front. Microbiol. 2022, 13, 1003692. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Cokcetin, N.N.; Burke, C.M.; Turnbull, L.; Liu, M.; Carter, D.A.; Whitchurch, C.B.; Harry, E.J. Honey can inhibit and eliminate biofilms produced by Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 18160. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Hensel, A.; Goycoolea, F.M. Chitosan/cyclodextrin surface-adsorbed naringenin-loaded nanocapsules enhance bacterial quorum quenching and anti-biofilm activities. Colloids Surf. B Biointerfaces 2022, 211, 112281. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Husain, F.M.; Zia, Q.; Ahmad, E.; Jamal, A.; Alaidarous, M.; Banawas, S.; Alam, M.M.; Alshehri, B.A.; Jameel, M. Anti-quorum sensing and anti-biofilm activity of zinc oxide nanospikes. ACS Omega 2020, 5, 32203–32215. [Google Scholar] [CrossRef]
- Kumar, S.; Paliya, B.S.; Singh, B.N. Superior inhibition of virulence and biofilm formation of Pseudomonas aeruginosa PAO1 by phyto-synthesized silver nanoparticles through anti-quorum sensing activity. Microb. Pathog. 2022, 170, 105678. [Google Scholar] [CrossRef]
- Bryers, J.D.; Jarvis, R.A.; Lebo, J.; Prudencio, A.; Kyriakides, T.R.; Uhrich, K. Biodegradation of poly (anhydride-esters) into non-steroidal anti-inflammatory drugs and their effect on Pseudomonas aeruginosa biofilms in vitro and on the foreign-body response in vivo. Biomaterials 2006, 27, 5039–5048. [Google Scholar] [CrossRef]
- Sarveswari, H.B.; Gupta, K.K.; Durai, R.; Solomon, A.P. Development of a smart pH-responsive nano-polymer drug, 2-methoxy-4-vinylphenol conjugate against the intestinal pathogen, Vibrio cholerae. Sci. Rep. 2023, 13, 1250. [Google Scholar] [CrossRef]
- Cavaleiro, E.; Duarte, A.S.; Esteves, A.C.; Correia, A.; Whitcombe, M.J.; Piletska, E.V.; Piletsky, S.A.; Chianella, I. Novel linear polymers able to inhibit bacterial quorum sensing. Macromol. Biosci. 2015, 15, 647–656. [Google Scholar] [CrossRef]
- Lidor, O.; Al-Quntar, A.; Pesci, E.; Steinberg, D. Mechanistic analysis of a synthetic inhibitor of the Pseudomonas aeruginosa LasI quorum-sensing signal synthase. Sci. Rep. 2015, 5, 16569. [Google Scholar] [CrossRef]
- Cui, C.; Song, S.; Yang, C.; Sun, X.; Huang, Y.; Li, K.; Zhao, S.; Zhang, Y.; Deng, Y. Disruption of quorum sensing and virulence in Burkholderia cenocepacia by a structural analogue of the cis-2-dodecenoic acid signal. Appl. Environ. Microbiol. 2019, 85, e00105–e00119. [Google Scholar] [CrossRef] [PubMed]
- Nicol, M.; Alexandre, S.; Luizet, J.-B.; Skogman, M.; Jouenne, T.; Salcedo, S.P.; Dé, E. Unsaturated fatty acids affect quorum sensing communication system and inhibit motility and biofilm formation of Acinetobacter baumannii. Int. J. Mol. Sci. 2018, 19, 214. [Google Scholar] [CrossRef] [PubMed]
- Hallan, S.S.; Marchetti, P.; Bortolotti, D.; Sguizzato, M.; Esposito, E.; Mariani, P.; Trapella, C.; Rizzo, R.; Cortesi, R. Design of nanosystems for the delivery of Quorum Sensing inhibitors: A preliminary study. Molecules 2020, 25, 5655. [Google Scholar] [CrossRef]
- Zafar, F.; Shahid, M.; Fatima, H.; Riaz, M.; Anjum, F.; Mushtaq, Z.; Zia, S.; Jahangir, M.M.; Aslam, M.A. Antibiofilm and Quorum Sensing Inhibition (QSI) Potential of Lagerstroemia speciosa Leaves Extract. Dose-Response 2022, 20, 15593258221132080. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, H.; Chu, X.; Lou, T.; Zhang, N.; Zhang, B.; Chu, W. Tea polyphenols inhibits biofilm formation, attenuates the quorum sensing-controlled virulence and enhances resistance to Klebsiella pneumoniae infection in Caenorhabditis elegans model. Microb. Pathog. 2020, 147, 104266. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Breslawec, A.P.; Liang, T.; Deng, Z.; Kuperman, L.L.; Yu, Q. Strategy to combat biofilms: A focus on biofilm dispersal enzymes. NPJ Biofilms Microbiomes 2023, 9, 63. [Google Scholar] [CrossRef]
- Tan, Y.; Ma, S.; Liu, C.; Yu, W.; Han, F. Enhancing the stability and antibiofilm activity of DspB by immobilization on carboxymethyl chitosan nanoparticles. Microbiol. Res. 2015, 178, 35–41. [Google Scholar] [CrossRef]
- Chen, K.-J.; Lee, C.-K. Twofold enhanced dispersin B activity by N-terminal fusion to silver-binding peptide for biofilm eradication. Int. J. Biol. Macromol. 2018, 118, 419–426. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Z.; Zeng, K.; Xia, Y.; Xu, W.; Wang, R.; Guo, J.; Xie, H. Functional immobilization of a biofilm-releasing glycoside hydrolase dispersin B on magnetic nanoparticles. Appl. Biochem. Biotechnol. 2022, 194, 737–747. [Google Scholar] [CrossRef]
- Darouiche, R.O.; Mansouri, M.D.; Gawande, P.V.; Madhyastha, S. Antimicrobial and antibiofilm efficacy of triclosan and DispersinB® combination. J. Antimicrob. Chemother. 2009, 64, 88–93. [Google Scholar] [CrossRef]
- Marcano, A.; Ba, O.; Thebault, P.; Crétois, R.; Marais, S.; Duncan, A.C. Elucidation of innovative antibiofilm materials. Colloids Surf. B Biointerfaces 2015, 136, 56–63. [Google Scholar] [CrossRef]
- Waryah, C.B.; Wells, K.; Ulluwishewa, D.; Chen-Tan, N.; Gogoi-Tiwari, J.; Ravensdale, J.; Costantino, P.; Gökçen, A.; Vilcinskas, A.; Wiesner, J. In vitro antimicrobial efficacy of tobramycin against Staphylococcus aureus biofilms in combination with or without DNase I and/or dispersin B: A preliminary investigation. Microb. Drug Resist. 2017, 23, 384–390. [Google Scholar] [CrossRef]
- Gawande, P.V.; Leung, K.P.; Madhyastha, S. Antibiofilm and antimicrobial efficacy of DispersinB®-KSL-W peptide-based wound gel against chronic wound infection associated bacteria. Curr. Microbiol. 2014, 68, 635–641. [Google Scholar] [CrossRef]
- Gawande, P.V.; Clinton, A.P.; LoVetri, K.; Yakandawala, N.; Rumbaugh, K.P.; Madhyastha, S. Antibiofilm efficacy of DispersinB® wound spray used in combination with a silver wound dressing. Microbiol. Insights 2014, 7, 9–13. [Google Scholar] [CrossRef]
- Little, D.J.; Pfoh, R.; Le Mauff, F.; Bamford, N.C.; Notte, C.; Baker, P.; Guragain, M.; Robinson, H.; Pier, G.B.; Nitz, M. PgaB orthologues contain a glycoside hydrolase domain that cleaves deacetylated poly-β (1, 6)-N-acetylglucosamine and can disrupt bacterial biofilms. PLoS Pathog. 2018, 14, e1006998. [Google Scholar] [CrossRef]
- Asker, D.; Awad, T.S.; Baker, P.; Howell, P.L.; Hatton, B.D. Non-eluting, surface-bound enzymes disrupt surface attachment of bacteria by continuous biofilm polysaccharide degradation. Biomaterials 2018, 167, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Asker, D.; Awad, T.S.; Raju, D.; Sanchez, H.; Lacdao, I.; Gilbert, S.; Sivarajah, P.; Andes, D.R.; Sheppard, D.C.; Howell, P.L. Preventing Pseudomonas aeruginosa biofilms on indwelling catheters by surface-bound enzymes. ACS Appl. Bio Mater. 2021, 4, 8248–8258. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.; Hill, P.J.; Snarr, B.D.; Alnabelseya, N.; Pestrak, M.J.; Lee, M.J.; Jennings, L.K.; Tam, J.; Melnyk, R.A.; Parsek, M.R. Exopolysaccharide biosynthetic glycoside hydrolases can be utilized to disrupt and prevent Pseudomonas aeruginosa biofilms. Sci. Adv. 2016, 2, e1501632. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.D.; Baker, P.; Bamford, N.C.; Sato, Y.; Liu, H.; Lehoux, M.; Gravelat, F.N.; Ostapska, H.; Baistrocchi, S.R.; Cerone, R.P. Microbial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc. Nati. Acad. Sci. USA 2017, 114, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Sunsunwal, S.; Gautam, V.; Singh, M.; Ramya, T. Biofilm inhibitory effect of alginate lyases on mucoid P. aeruginosa from a cystic fibrosis patient. Biochem. Biophys. Rep. 2021, 26, 101028. [Google Scholar] [CrossRef]
- Daboor, S.M.; Rohde, J.R.; Cheng, Z. Disruption of the extracellular polymeric network of Pseudomonas aeruginosa biofilms by alginate lyase enhances pathogen eradication by antibiotics. J. Cyst. Fibros. 2021, 20, 264–270. [Google Scholar] [CrossRef]
- Alkawash, M.A.; Soothill, J.S.; Schiller, N.L. Alginate lyase enhances antibiotic killing of mucoid Pseudomonas aeruginosa in biofilms. APMIS 2006, 114, 131–138. [Google Scholar] [CrossRef]
- Patel, K.K.; Tripathi, M.; Pandey, N.; Agrawal, A.K.; Gade, S.; Anjum, M.M.; Tilak, R.; Singh, S. Alginate lyase immobilized chitosan nanoparticles of ciprofloxacin for the improved antimicrobial activity against the biofilm associated mucoid P. aeruginosa infection in cystic fibrosis. Int. J. Pharm. 2019, 563, 30–42. [Google Scholar] [CrossRef]
- Islan, G.A.; Dini, C.; Bartel, L.C.; Bolzán, A.D.; Castro, G.R. Characterization of smart auto-degradative hydrogel matrix containing alginate lyase to enhance levofloxacin delivery against bacterial biofilms. Int. J. Pharm. 2015, 496, 953–964. [Google Scholar] [CrossRef]
- Charęza, M.; Przygrodzka, K.; Żywicka, A.; Grygorcewicz, B.; Sobolewski, P.; Mozia, S.; Śmiglak, M.; Drozd, R. Enhancement of Inhibition of the Pseudomonas sp. Biofilm Formation on Bacterial Cellulose-Based Wound Dressing by the Combined Action of Alginate Lyase and Gentamicin. Int. J. Mol. Sci. 2023, 24, 4740. [Google Scholar] [CrossRef]
- Tetz, G.V.; Artemenko, N.K.; Tetz, V.V. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 2009, 53, 1204–1209. [Google Scholar] [CrossRef]
- Sharma, K.; Pagedar Singh, A. Antibiofilm effect of DNase against single and mixed species biofilm. Foods 2018, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Pakkulnan, R.; Thonglao, N.; Chareonsudjai, S. DNase I and chitosan enhance efficacy of ceftazidime to eradicate Burkholderia pseudomallei biofilm cells. Sci. Rep. 2023, 13, 1059. [Google Scholar] [CrossRef] [PubMed]
- Baelo, A.; Levato, R.; Julian, E.; Crespo, A.; Astola, J.; Gavalda, J.; Engel, E.; Mateos-Timoneda, M.A.; Torrents, E. Disassembling bacterial extracellular matrix with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections. J. Control. Release 2015, 209, 150–158. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Y.; Su, W.; Chai, J.; Xu, L.; Cao, J.; Liu, Y. Encapsulated DNase improving the killing efficiency of antibiotics in staphylococcal biofilms. J. Mater. Chem. B 2020, 8, 4395–4401. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Agrawal, A.K.; Anjum, M.M.; Tripathi, M.; Pandey, N.; Bhattacharya, S.; Tilak, R.; Singh, S. DNase-I functionalization of ciprofloxacin-loaded chitosan nanoparticles overcomes the biofilm-mediated resistance of Pseudomonas aeruginosa. Appl. Nanosci. 2020, 10, 563–575. [Google Scholar] [CrossRef]
- Karygianni, L.; Attin, T.; Thurnheer, T. Combined DNase and proteinase treatment interferes with composition and structural integrity of multispecies oral biofilms. J. Clin. Med. 2020, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Jee, S.-C.; Kim, M.; Sung, J.-S.; Kadam, A.A. Efficient biofilms eradication by enzymatic-cocktail of pancreatic protease type-I and bacterial α-amylase. Polymers 2020, 12, 3032. [Google Scholar] [CrossRef]
- Weldrick, P.J.; Hardman, M.J.; Paunov, V.N. Smart active antibiotic nanocarriers with protease surface functionality can overcome biofilms of resistant bacteria. Mater. Chem. Front. 2021, 5, 961–972. [Google Scholar] [CrossRef]
- Weldrick, P.J.; Hardman, M.J.; Paunov, V.N. Enhanced clearing of wound-related pathogenic bacterial biofilms using protease-functionalized antibiotic nanocarriers. ACS Appl. Mater. Interfaces 2019, 11, 43902–43919. [Google Scholar] [CrossRef]
- Little, D.J. Modification and Translocation of the Biofilm Exopolysaccharide Poly-β (1, 6)-N-acetyl-D-glucosamine. Ph.D. Thesis, University of Toronto, Toronto, ON, Canada, 2015. [Google Scholar]
- Breslawec, A.P.; Wang, S.; Li, C.; Poulin, M.B. Anionic amino acids support hydrolysis of poly-β-(1, 6)-N-acetylglucosamine exopolysaccharides by the biofilm dispersing glycosidase Dispersin B. J. Biol. Chem. 2021, 296, 100203. [Google Scholar] [CrossRef]
- Thorn, C.R.; Howell, P.L.; Wozniak, D.J.; Prestidge, C.A.; Thomas, N. Enhancing the therapeutic use of biofilm-dispersing enzymes with smart drug delivery systems. Adv. Drug Deliv. Rev. 2021, 179, 113916. [Google Scholar] [CrossRef]
- Ghalsasi, V.; Sourjik, V. Engineering Escherichia coli to disrupt poly-N-acetylglucosamine containing bacterial biofilms. Curr. Synth. Syst. Biol. 2016, 4, 2332-0737. [Google Scholar] [CrossRef]
- Abdelkader, J.; Alelyani, M.; Alashban, Y.; Alghamdi, S.A.; Bakkour, Y. Modification of Dispersin B with Cyclodextrin-Ciprofloxacin Derivatives for Treating Staphylococcal. Molecules 2023, 28, 5311. [Google Scholar] [CrossRef]
- Serrera, A.; del Pozo, J.; Martinez, A.; Alonso, M.; Gonzalez, R.; Leiva, J.; Vergara, M.; Lasa, I. P1786 Dispersin B therapy of Staphylococcus aureus experimental port-related bloodstream infection. Int. J. Antimicrob. Agents 2007, 29, S508. [Google Scholar] [CrossRef]
- Forman, A.; Pfoh, R.; Eddenden, A.; Howell, P.L.; Nitz, M. Synthesis of defined mono-de-N-acetylated β-(1→ 6)-N-acetyl-D-glucosamine oligosaccharides to characterize PgaB hydrolase activity. Org. Biomol. Chem. 2019, 17, 9456–9466. [Google Scholar] [CrossRef]
- Blanco-Cabra, N.; Paetzold, B.; Ferrar, T.; Mazzolini, R.; Torrents, E.; Serrano, L.; Luch-Senar, M. Characterization of different alginate lyases for dissolving Pseudomonas aeruginosa biofilms. Sci. Rep. 2020, 10, 9390. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, H. Alginate lyase: Review of major sources and classification, properties, structure-function analysis and applications. Bioengineered 2015, 6, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Suntres, Z.E.; Omri, A. Importance of DNase and alginate lyase for enhancing free and liposome encapsulated aminoglycoside activity against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2009, 64, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Mrsny, R.; Lazazzera, B.; Daugherty, A.; Schiller, N.; Patapoff, T. Addition of a bacterial alginate lyase to purulent CF sputum in vitro can result in the disruption of alginate and modification of sputum viscoelasticity. Pulm. Pharmacol. 1994, 7, 357–366. [Google Scholar] [CrossRef]
- Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics 2020, 10, 3. [Google Scholar] [CrossRef]
- Sikdar, R.; Elias, M. Quorum quenching enzymes and their effects on virulence, biofilm, and microbiomes: A review of recent advances. Expert Rev. Anti-Infect. Ther. 2020, 18, 1221–1233. [Google Scholar] [CrossRef]
- Daboor, S.M.; Raudonis, R.; Cheng, Z. Characterizations of the viability and gene expression of dispersal cells from Pseudomonas aeruginosa biofilms released by alginate lyase and tobramycin. PLoS ONE 2021, 16, e0258950. [Google Scholar] [CrossRef]
- Baker, P.; Whitfield, G.B.; Hill, P.J.; Little, D.J.; Pestrak, M.J.; Robinson, H.; Wozniak, D.J.; Howell, P.L. Characterization of the Pseudomonas aeruginosa glycoside hydrolase PslG reveals that its levels are critical for Psl polysaccharide biosynthesis and biofilm formation. J. Biol. Chem. 2015, 290, 28374–28387. [Google Scholar] [CrossRef] [PubMed]
- Pestrak, M.J.; Baker, P.; Dellos-Nolan, S.; Hill, P.J.; Passos da Silva, D.; Silver, H.; Lacdao, I.; Raju, D.; Parsek, M.R.; Wozniak, D.J. Treatment with the Pseudomonas aeruginosa glycoside hydrolase PslG combats wound infection by improving antibiotic efficacy and host innate immune activity. Antimicrob. Agents Chemother. 2019, 63, e00234-19. [Google Scholar] [CrossRef] [PubMed]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B.; LoVetri, K.; Cardona, S.T.; Madhyastha, S.; Sadovskaya, I.; Jabbouri, S.; Izano, E.A. Recombinant human DNase I decreases biofilm and increases antimicrobial susceptibility in staphylococci. J. Antibiot. 2012, 65, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Shire, S.J.; Scherer, T.M. The pharmaceutical development of rhDNase (Dornase Alpha) for the treatment of cystic fibrosis. In Mucosal Delivery of Biopharmaceuticals: Biology, Challenges and Strategies; Springer: Berlin/Heidelberg, Germany, 2014; pp. 437–459. [Google Scholar]
- Fanaei Pirlar, R.; Emaneini, M.; Beigverdi, R.; Banar, M.; van Leeuwen, W.B.; Jabalameli, F. Combinatorial effects of antibiotics and enzymes against dual-species Staphylococcus aureus and Pseudomonas aeruginosa biofilms in the wound-like medium. PLoS ONE 2020, 15, e0235093. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Surekha, D.B.; Tripathi, M.; Anjum, M.M.; Muthu, M.; Tilak, R.; Agrawal, A.K.; Singh, S. Antibiofilm potential of silver sulfadiazine-loaded nanoparticle formulations: A study on the effect of DNase-I on microbial biofilm and wound healing activity. Mol. Pharm. 2019, 16, 3916–3925. [Google Scholar] [CrossRef] [PubMed]
- Osman, R.; Kan, P.L.; Awad, G.; Mortada, N.; Abd-Elhameed, E.-S.; Alpar, O. Enhanced properties of discrete pulmonary deoxyribonuclease I (DNaseI) loaded PLGA nanoparticles during encapsulation and activity determination. Int. J. Pharm. 2011, 408, 257–265. [Google Scholar] [CrossRef]
- Kaplan, J.Á. Biofilm dispersal: Mechanisms, clinical implications, and potential therapeutic uses. J. Dent. Res. 2010, 89, 205–218. [Google Scholar] [CrossRef]
- Jiang, Y.; Geng, M.; Bai, L. Targeting biofilms therapy: Current research strategies and development hurdles. Microorganisms 2020, 8, 1222. [Google Scholar] [CrossRef]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar] [CrossRef]
- Shukla, S.K.; Rao, T.S. Staphylococcus aureus biofilm removal by targeting biofilm-associated extracellular proteins. Indian J. Med. Res. 2017, 146, S1. [Google Scholar]
- Marx, C.; Gardner, S.; Harman, R.M.; Van de Walle, G.R. The mesenchymal stromal cell secretome impairs methicillin-resistant Staphylococcus aureus biofilms via cysteine protease activity in the equine model. Stem Cells Transl. Med. 2020, 9, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Diab, R.; Khameneh, B.; Joubert, O.; Duval, R. Insights in nanoparticle-bacterium interactions: New frontiers to bypass bacterial resistance to antibiotics. Curr. Pharm. Des. 2015, 21, 4095–4105. [Google Scholar] [CrossRef] [PubMed]
- Ikuma, K.; Decho, A.W.; Lau, B.L. When nanoparticles meet biofilms—Interactions guiding the environmental fate and accumulation of nanoparticles. Front. Microbiol. 2015, 6, 591. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, S.; Wang, J.; Chen, Q. A review on polymer and lipid-based nanocarriers and its application to nano-pharmaceutical and food-based systems. Front. Nutr. 2021, 8, 783831. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, M.A.; Taleuzzaman, M.; Kala, C.; Gilani, S.J. Advancements in polymer and lipid-based nanotherapeutics for cancer drug targeting. Curr. Pharm. Des. 2020, 26, 5119–5127. [Google Scholar] [CrossRef]
- Baptista, P.V.; McCusker, M.P.; Carvalho, A.; Ferreira, D.A.; Mohan, N.M.; Martins, M.; Fernandes, A.R. Nano-strategies to fight multidrug resistant bacteria—“A Battle of the Titans”. Front. Microbiol. 2018, 9, 1441. [Google Scholar] [CrossRef] [PubMed]
- Henostroza, M.A.B.; Tavares, G.D.; Yukuyama, M.N.; De Souza, A.; Barbosa, E.J.; Avino, V.C.; dos Santos Neto, E.; Lourenço, F.R.; Löbenberg, R.; Bou-Chacra, N.A. Antibiotic-loaded lipid-based nanocarrier: A promising strategy to overcome bacterial infection. Int. J. Pharm. 2022, 621, 121782. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Adhipandito, C.F.; Cheung, S.H.; Lin, Y.H.; Wu, S.H. Atypical renal clearance of nanoparticles larger than the kidney filtration threshold. Int. J. Mol. Sci. 2021, 22, 11182. [Google Scholar] [CrossRef]
- Selvamani, V. Stability studies on nanomaterials used in drugs. In Characterization and Biology of Nanomaterials for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2019; pp. 425–444. [Google Scholar]
- Joshi, A.S.; Singh, P.; Mijakovic, I. Interactions of gold and silver nanoparticles with bacterial biofilms: Molecular interactions behind inhibition and resistance. Int. J. Mol. Sci. 2020, 21, 7658. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, L.; Su, L.; van der Mei, H.C.; Jutte, P.C.; Ren, Y.; Busscher, H.J. Nanotechnology-based antimicrobials and delivery systems for biofilm-infection control. Chem. Soc. Rev. 2019, 48, 428–446. [Google Scholar] [CrossRef]
- Sankaran, J.; Tan, N.J.; But, K.P.; Cohen, Y.; Rice, S.A.; Wohland, T. Single microcolony diffusion analysis in Pseudomonas aeruginosa biofilms. NPJ Biofilms Microbiomes 2019, 5, 35. [Google Scholar] [CrossRef]
- Ong, T.H.; Chitra, E.; Ramamurthy, S.; Ling, C.C.S.; Ambu, S.P.; Davamani, F. Cationic chitosan-propolis nanoparticles alter the zeta potential of S. epidermidis, inhibit biofilm formation by modulating gene expression and exhibit synergism with antibiotics. PLoS ONE 2019, 14, e0213079. [Google Scholar]
- Ferreira, M.; Pinto, S.N.; Aires-da-Silva, F.; Bettencourt, A.; Aguiar, S.I.; Gaspar, M.M. Liposomes as a Nanoplatform to Improve the Delivery of Antibiotics into Staphylococcus aureus Biofilms. Pharmaceutics 2021, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Alhariri, M.; Majrashi, M.A.; Bahkali, A.H.; Almajed, F.S.; Azghani, A.O.; Khiyami, M.A.; Alyamani, E.J.; Aljohani, S.M.; Halwani, M.A. Efficacy of neutral and negatively charged liposome-loaded gentamicin on planktonic bacteria and biofilm communities. Int. J. Nanomed. 2017, 12, 6949–6961. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Dkhar, D.S.; Kumari, R.; Mahapatra, S.; Dubey, V.K.; Chandra, P. Lipid based nanocarriers: Production techniques, concepts, and commercialization aspect. J. Drug Deliv. Sci. Technol. 2022, 74, 103526. [Google Scholar] [CrossRef]
- Tan, C.; Wang, J.; Sun, B. Biopolymer-liposome hybrid systems for controlled delivery of bioactive compounds: Recent advances. Biotechnol. Adv. 2021, 48, 107727. [Google Scholar] [CrossRef]
- Raemdonck, K.; Braeckmans, K.; Demeester, J.; De Smedt, S.C. Merging the best of both worlds: Hybrid lipid-enveloped matrix nanocomposites in drug delivery. Chem. Soc. Rev. 2014, 43, 444–472. [Google Scholar] [CrossRef]
- Esposto, B.S.; Jauregi, P.; Tapia-Blácido, D.R.; Martelli-Tosi, M. Liposomes vs. chitosomes: Encapsulating food bioactives. Trends Food Sci. Technol. 2021, 108, 40–48. [Google Scholar] [CrossRef]
- Pinilla, C.M.B.; Lopes, N.A.; Brandelli, A. Lipid-based nanostructures for the delivery of natural antimicrobials. Molecules 2021, 26, 3587. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, Z.; Zhang, P.; Bai, H.; Sun, Y.; Duan, J.; Mu, H. Lysozyme associated liposomal gentamicin inhibits bacterial biofilm. Int. J. Mol. Sci. 2017, 18, 784. [Google Scholar] [CrossRef] [PubMed]
- Tseng, B.S.; Zhang, W.; Harrison, J.J.; Quach, T.P.; Song, J.L.; Penterman, J.; Singh, P.K.; Chopp, D.L.; Packman, A.I.; Parsek, M.R. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ. Microbiol. 2013, 15, 2865–2878. [Google Scholar] [CrossRef]
- Yoshimoto, M. Stabilization of enzymes through encapsulation in liposomes. In Enzyme Stabilization and Immobilization: Methods and Protocols; Humana: New York, NY, USA, 2017; pp. 9–18. [Google Scholar]
- Fang, J.-Y.; Chou, W.-L.; Lin, C.-F.; Sung, C.T.; Alalaiwe, A.; Yang, S.-C. Facile biofilm penetration of cationic liposomes loaded with DNase I/Proteinase K to eradicate Cutibacterium acnes for treating cutaneous and catheter infections. Int. J. Nanomed. 2021, 16, 8121–8138. [Google Scholar] [CrossRef]
- Bandara, H.; Herpin, M.; Kolacny, D., Jr.; Harb, A.; Romanovicz, D.; Smyth, H. Incorporation of farnesol significantly increases the efficacy of liposomal ciprofloxacin against Pseudomonas aeruginosa biofilms in vitro. Mol. Pharm. 2016, 13, 2760–2770. [Google Scholar] [CrossRef]
- Alarfaj, R.E.; Alkhulaifi, M.M.; Al-Fahad, A.J.; Aljihani, S.; Yassin, A.E.B.; Alghoribi, M.F.; Halwani, M.A. Antibacterial efficacy of liposomal formulations containing tobramycin and N-acetylcysteine against tobramycin-resistant Escherichia coli, Klebsiella pneumoniae, and acinetobacter baumannii. Pharmaceutics 2022, 14, 130. [Google Scholar] [CrossRef]
- Aljihani, S.A.; Alehaideb, Z.; Alarfaj, R.E.; Alghoribi, M.F.; Akiel, M.A.; Alenazi, T.H.; Al-Fahad, A.J.; Al Tamimi, S.M.; Albakr, T.M.; Alshehri, A.; et al. Enhancing azithromycin antibacterial activity by encapsulation in liposomes/liposomal-N-acetylcysteine formulations against resistant clinical strains of Escherichia coli. Saudi J. Biol. Sci. 2020, 27, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Tonguc-Altin, K.; Sandalli, N.; Duman, G.; Selvi-Kuvvetli, S.; Topcuoglu, N.; Kulekci, G. Development of novel formulations containing Lysozyme and Lactoferrin and evaluation of antibacterial effects on Mutans Streptococci and Lactobacilli. Arch. Oral Biol. 2015, 60, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.V.; Nirwane, A.M.; Belubbi, T.; Nagarsenker, M.S. Pulmonary delivery of synergistic combination of fluoroquinolone antibiotic complemented with proteolytic enzyme: A novel antimicrobial and antibiofilm strategy. Nanomedicine 2017, 13, 2371–2384. [Google Scholar] [CrossRef]
- Rao, Y.; Sun, Y.; Li, P.; Xu, M.; Chen, X.; Wang, Y.; Chen, Y.; Deng, X.; Yu, S.; Hu, H. Hypoxia-sensitive adjuvant loaded liposomes enhance the antimicrobial activity of azithromycin via phospholipase-triggered releasing for Pseudomonas aeruginosa biofilms eradication. Int. J. Pharm. 2022, 623, 121910. [Google Scholar] [CrossRef]
- Alhariri, M.; Omri, A. Efficacy of Liposomal Bismuth-Ethanedithiol-Loaded Tobramycin after Intratracheal Administration in Rats with Pulmonary Pseudomonas aeruginosa Infection. Antimicrob. Agents Chemother. 2013, 57, 569–578. [Google Scholar] [CrossRef]
- Alipour, M.; Dorval, C.; Suntres, Z.E.; Omri, A. Bismuth-ethanedithiol incorporated in a liposome-loaded tobramycin formulation modulates the alginate levels in mucoid Pseudomonas aeruginosa. J. Pharm. Pharmacol. 2011, 63, 999–1007. [Google Scholar] [CrossRef]
- Thorn, C.R.; Prestidge, C.A.; Boyd, B.J.; Thomas, N. Pseudomonas infection responsive liquid crystals for glycoside hydrolase and antibiotic combination. ACS Appl. Bio Mater. 2018, 1, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.K.; Murgia, X.; De Rossi, C.; Christmann, R.; Hüfner de Mello Martins, A.G.; Koch, M.; Andreas, A.; Herrmann, J.; Müller, R.; Empting, M. Cover Picture: Squalenyl Hydrogen Sulfate Nanoparticles for Simultaneous Delivery of Tobramycin and an Alkylquinolone Quorum Sensing Inhibitor Enable the Eradication of P. aeruginosa Biofilm Infections. Angew. Chem. Int. Ed. 2020, 59, 10201. [Google Scholar]
- Islan, G.A.; Tornello, P.C.; Abraham, G.A.; Duran, N.; Castro, G.R. Smart lipid nanoparticles containing levofloxacin and DNase for lung delivery. Design and characterization. Colloids Surf. B Biointerfaces 2016, 143, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Akhtari, H.; Fazly Bazzaz, B.S.; Golmohammadzadeh, S.; Movaffagh, J.; Soheili, V.; Khameneh, B. Rifampin and cis-2-decenoic acid co-entrapment in solid lipid nanoparticles as an efficient nano-system with potent anti-biofilm activities. J. Pharm. Innov. 2021, 16, 293–301. [Google Scholar] [CrossRef]
- Anjum, M.M.; Patel, K.K.; Dehari, D.; Pandey, N.; Tilak, R.; Agrawal, A.K.; Singh, S. Anacardic acid encapsulated solid lipid nanoparticles for Staphylococcus aureus biofilm therapy: Chitosan and DNase coating improves antimicrobial activity. Drug Deliv. Transl. Res. 2021, 11, 305–317. [Google Scholar] [CrossRef]
- Pinto, R.M.; Monteiro, C.; Costa Lima, S.A.; Casal, S.; Van Dijck, P.; Martins, M.C.L.; Nunes, C.; Reis, S. N-acetyl-l-cysteine-loaded nanosystems as a promising therapeutic approach toward the eradication of Pseudomonas aeruginosa biofilms. ACS Appl. Mater. Interfaces 2021, 13, 42329–42343. [Google Scholar] [CrossRef]
- Quatrin, P.M.; Verdi, C.M.; de Souza, M.E.; de Godoi, S.N.; Klein, B.; Gundel, A.; Wagner, R.; de Almeida Vaucher, R.; Ourique, A.F.; Santos, R.C.V. Antimicrobial and antibiofilm activities of nanoemulsions containing Eucalyptus globulus oil against Pseudomonas aeruginosa and Candida spp. Microb. Pathog. 2017, 112, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.Y.; Kee, L.T.; Al-Masawa, M.E.; Lee, Q.H.; Subramaniam, T.; Kok, D.; Ng, M.H.; Law, J.X. Scalable production of extracellular vesicles and its therapeutic values: A review. Int. J. Mol. Sci. 2022, 23, 7986. [Google Scholar] [CrossRef]
- Lee, K.W.; Nguyen, T.-H.; Hanley, T.; Boyd, B.J. Nanostructure of liquid crystalline matrix determines in vitro sustained release and in vivo oral absorption kinetics for hydrophilic model drugs. Int. J. Pharm. 2009, 365, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Fernandez, G.; Blanco-Fernandez, B.; Fernández-Ferreiro, A.; Otero-Espinar, F.J. Lipidic lyotropic liquid crystals: Insights on biomedical applications. Adv. Colloid Interface Sci. 2023, 313, 102867. [Google Scholar] [CrossRef] [PubMed]
- Waghule, T.; Patil, S.; Rapalli, V.K.; Girdhar, V.; Gorantla, S.; Kumar Dubey, S.; Saha, R.N.; Singhvi, G. Improved skin-permeated diclofenac-loaded lyotropic liquid crystal nanoparticles: QbD-driven industrial feasible process and assessment of skin deposition. Liq. Cryst. 2021, 48, 991–1009. [Google Scholar] [CrossRef]
- Huang, Y.; Gui, S. Factors affecting the structure of lyotropic liquid crystals and the correlation between structure and drug diffusion. RSC Adv. 2018, 8, 6978–6987. [Google Scholar] [CrossRef]
- Shetty, S.; Shetty, S. Cubosome-based cosmeceuticals: A breakthrough in skincare. Drug Discov. Today 2023, 28, 103623. [Google Scholar] [CrossRef]
- Jampilek, J.; Kralova, K. Advances in nanostructures for antimicrobial therapy. Materials 2022, 15, 2388. [Google Scholar] [CrossRef]
- Rajak, P.; Nath, L.; Bhuyan, B. Liquid Crystals: An Approach in Drug Delivery. Indian J. Pharm. Sci. 2019, 81, 11–21. [Google Scholar] [CrossRef]
- Thorn, C.R.; Clulow, A.J.; Boyd, B.J.; Prestidge, C.A.; Thomas, N. Bacterial lipase triggers the release of antibiotics from digestible liquid crystal nanoparticles. J. Control. Release 2020, 319, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Mao, Z.; Ran, F.; Sun, J.; Zhang, J.; Chai, G.; Wang, J. Nanotechnology-Based Drug Delivery Systems to Control Bacterial-Biofilm-Associated Lung Infections. Pharmaceutics 2023, 15, 2582. [Google Scholar] [CrossRef]
- Leu, J.S.; Teoh, J.J.; Ling, A.L.; Chong, J.; Loo, Y.S.; Mat Azmi, I.D.; Zahid, N.I.; Bose, R.J.; Madheswaran, T. Recent Advances in the Development of Liquid Crystalline Nanoparticles as Drug Delivery Systems. Pharmaceutics 2023, 15, 1421. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, W.; Mäder, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2012, 64, 83–101. [Google Scholar] [CrossRef]
- Rehman, A.U.; Akram, S.; Seralin, A.; Vandamme, T.; Anton, N. Lipid nanocarriers: Formulation, properties, and applications. In Smart Nanocontainers; Elsevier: Amsterdam, The Netherlands, 2020; pp. 355–382. [Google Scholar]
- Luan, L.; Chi, Z.; Liu, C. Chinese white wax solid lipid nanoparticles as a novel nanocarrier of curcumin for inhibiting the formation of Staphylococcus aureus biofilms. Nanomaterials 2019, 9, 763. [Google Scholar] [CrossRef] [PubMed]
- Nafee, N.; Husari, A.; Maurer, C.K.; Lu, C.; de Rossi, C.; Steinbach, A.; Hartmann, R.W.; Lehr, C.-M.; Schneider, M. Antibiotic-free nanotherapeutics: Ultra-small, mucus-penetrating solid lipid nanoparticles enhance the pulmonary delivery and anti-virulence efficacy of novel quorum sensing inhibitors. J. Control. Release 2014, 192, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured lipid carriers for delivery of chemotherapeutics: A review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.-H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef]
- Akhavan, S.; Assadpour, E.; Katouzian, I.; Jafari, S.M. Lipid nano scale cargos for the protection and delivery of food bioactive ingredients and nutraceuticals. Trends Food Sci. Technol. 2018, 74, 132–146. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, W.; Lei, E.; Yang, A.; Li, Y.; Wen, K.; Wang, M.; Li, L.; Chen, Z.; Zhou, C. Cooperative Membrane Damage as a Mechanism for Pentamidine–Antibiotic Mutual Sensitization. ACS Chem. Biol. 2022, 17, 3178–3190. [Google Scholar] [CrossRef]
- Vishwakarma, V. Impact of environmental biofilms: Industrial components and its remediation. J. Basic Microbiol. 2020, 60, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Rather, M.A.; Gupta, K.; Bardhan, P.; Borah, M.; Sarkar, A.; Eldiehy, K.S.; Bhuyan, S.; Mandal, M. Microbial biofilm: A matter of grave concern for human health and food industry. J. Basic Microbiol. 2021, 61, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.L.; Li, Q.-l.; Chu, C.-H.; Lo, E.-M.; Samaranayake, L.P. Antibacterial effects of silver diamine fluoride on multi-species cariogenic biofilm on caries. Ann. Clin. Microbiol. Antimicrob. 2013, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Quan, X.; Si, X.; Wang, X. A small molecule norspermidine in combination with silver ion enhances dispersal and disinfection of multi-species wastewater biofilms. Appl. Microbiol. Biotechnol. 2016, 100, 5619–5629. [Google Scholar] [CrossRef]
- Ioannidis, K.; Niazi, S.; Mylonas, P.; Mannocci, F.; Deb, S. The synthesis of nano silver-graphene oxide system and its efficacy against endodontic biofilms using a novel tooth model. Dent. Mater. 2019, 35, 1614–1629. [Google Scholar] [CrossRef]
- Hawas, S.; Verderosa, A.D.; Totsika, M. Combination therapies for biofilm inhibition and eradication: A comparative review of laboratory and preclinical studies. Front. Cell. Infect. Microbiol. 2022, 12, 850030. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Bashiri, S.; Yuan, Y.; Ziora, Z.M.; Nabil, O.; Masuda, K.; Khongkow, M.; Rimsueb, N.; Cabral, H.; Ruktanonchai, U. Antimicrobial activity enhancers: Towards smart delivery of antimicrobial agents. Antibiotics 2022, 11, 412. [Google Scholar] [CrossRef]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid–polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef]
- Lourenço, T.G.B.; Heller, D.; Silva-Boghossian, C.M.; Cotton, S.L.; Paster, B.J.; Colombo, A.P.V. Microbial signature profiles of periodontally healthy and diseased patients. J. Clin. Periodontol. 2014, 41, 1027–1036. [Google Scholar] [CrossRef]








| Quorum Sensing Inhibitors/Quorum Quenchers | Targeted Component | Tested Organism | Result | Ref. |
|---|---|---|---|---|
| 3-amino-7-chloro-2-nonylquinazolin-4(3H)-one (ACNQ) | Effective inhibitor of the PqsR receptor | P. aeruginosa | Complete eradication of 24 h P. aeruginosa biofilm infections. | [61] |
| (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone (furanone C-30) | afeI and afeR genes in Acidithiobacillus ferrooxidans | Acidithiobacillus ferrooxidans | Successfully inhibited the EPS production and hence biofilm formation and significantly downregulated the gene expression involved in biofilm growth. | [90] |
| Trans-cinnamaldehyde | E. faecalis biofilm cells | E. faecalis biofilm | No significant increase in E. faecalis biofilm metabolic activity and no significant reduction in cell viability after long-term treatment. | [91] |
| Trans-cinnamaldehyde | S. mutans UA159 genes | S. mutans | Significantly reduced plaque formation in a rat carrier model. | [92] |
| AiiA lactonase enzyme (produced by engineered T7 bacteriophage) | 3-oxo-C8-HSL (AHL) 3-oxo-C12-HSL (AHL) | A. tumefaciens P. aeruginosa | Degradation of 3-oxo-C8-HSL produced by A. tumefaciens KYC6 and degradation of 3-oxo-C12-HSL produced by P. aeruginosa. | [93] |
| Β-Sitosterol glucoside | AL-3 | E. coli | Complete inhibition of E. coli O157:H7 motility ≥2-fold reduction of E. coli biofilm formation. | [94] |
| Quercetin | P. aeruginosa PAO1 Las and Rhl QS circuits | P. aeruginosa | Significantly reduced PAO1 biofilm formation (50% reduction) and inhibited PAO1 adhesion. Significantly reduced PAO1 QS gene expression (lasI, lasR, rhlI, rhlR) | [95] |
| Echinatin | AI-2 | E. coli clinical isolated strains | Reduced biofilm EPS production and virulence factors. | [96] |
| Ajoene | Gac/Rsm QS circuit | P. aeruginosa and S. aureus | Reduced biofilm mass and sRNA expression. | [77] |
| Honey | P. aeruginosa biofilm | P. aeruginosa | Effective inhibition and eradication of P. aeruginosa biofilm. Significant reduction in living bacteria cells in P. aeruginosa biofilms at a sub-inhibitory concentration. | [97] |
| Naringenin | AHL uptake pathway | E. coli | Significant reduction in biofilm formation compared to un-formulated solution. | [98] |
| Zinc oxide nano spikes | N-acyl-homoserine lactone P. aeruginosa las and psq QS circuits | P. aeruginosa | Significant reduction in the production of virulent factors, and inhibited up to 80% of biofilm formation at half MIC. | [99] |
| Silver nanoparticles | P. aeruginosa las, rhl and psq QS circuits | P. aeruginosa | Reduction in PAO1 virulence gene expression, swarming activity, and biofilm formation. | [100] |
| Salicylic acid | P. aeruginosa las QS circuit | P. aeruginosa | Significant biofilm inhibition after 48 h. | [101] |
| 2-methoxy-4-vinyl phenol (2M4VP) | LuxO active site | V. cholerae | Inhibited up to 50% of biofilm formation and repression of virulence genes. | [102] |
| Linear copolymers (pMAA25-co-pMMA75 and pIA25-co-pMMA75) | 3-oxo-C6-HSL, C4-HSL, C6-HSL | V. fisheri, A. hydrophilia | Reduced V. fisheri bioluminescence and A. hydrophilia biofilm formation. | [103] |
| (z)-5-octylidenethiazolidine-2,4-dione (TZD-C8) | LasI | P. aeruginosa | 70% biofilm biomass reduction at MIC. | [104] |
| Cis-14-methylpantane-2-enoic acid | RpfFBc | Burkholderia sp. | Significant inhibition of biofilm formation and decreased QS-mediated virulence factors. | [105] |
| Palmitoleic acid Myristoleic acid | abaR | A. baumannii reference and clinical strains | Dispersed 24 h biofilm and inhibited 40% of biofilm formation. | [106] |
| CDC PF | LasR transcription regulator | P. aeruginosa | Formulated QSI prolonged biofilm treatment for 48 h, resulting in ≥20% inhibition. | [107] |
| L. speciosa extracts | QS genes | Sinusitis bacteria isolates | Significant antibiofilm activity, ≥ 50% biofilm inhibition, but ineffective against S. aureus. | [108] |
| Tea polyphenols (TP) | QS virulence | K. pneumonia and C. violaceum | Inhibited 23.7% of biofilm formation at half-MIC of TP, reduced C. elegans death (26.7%) at half-MIC of TP. | [109] |
| EPS-Degrading Enzyme | Targeted Component | Carrier System with/without Combined Antimicrobial | Test Organism | Result | Ref. |
|---|---|---|---|---|---|
| DspB | GlcNAc-(β-1,6)- GlcNAc | DspB loaded on Carboxymethyl chitosan nanoparticles | A. actinomycetemcomitan, Staphylococcus aureus and Staphylococcus epidermidis | Improved enzyme reusability and stability as well as enhanced biofilm inhibition and eradication efficacy compared to non-formulated solution. | [112] |
| DspB | - | Silver nanoparticles fused with DspB | Staphylococcus epidermidis | Enhance the biofilm eradication potential by 2-fold. | [113] |
| DspB | - | Fusion of DspB with magnetoreceptor protein and subsequently loaded onto Fe3O4@SiO2 nanoparticles | Staphylococcus sp., Staphylococcus aureus, Pseudomonas Putida, Bacillus Cereus | Enzymatic killing by DspB was increased with 40–60% biofilm removal. | [114] |
| DspB | - | DspB + triclosan coated on vascular catheters | Staphylococcus aureus | Enhanced the biofilm eradication biofilms compared with control, DspB alone, or triclosan alone, thus demonstrating synergistic anti-biofilm activity of the combination treatment. | [115] |
| DspB | - | DspB loaded onto Polyhydroxyalkanoate asymmetrical membranes | Staphylococcus epidermidis | Weakly inhibited the biofilm formation but effectively disrupted the preformed biofilms. | [116] |
| DspB DNase I | - | DspB + DNase I + Tobramycin | S. aureus | Combined treatment of tobramycin with either DspB or DNase I decreased bacterial load in S. aureus biofilms by 7500-fold and 8780-fold respectively, while tobramycin alone reduced cell numbers by only 40-fold. Combined treatment with both enzymes did not significantly enhance the tobramycin efficacy. | [117] |
| DspB | - | DspB + KSL-W peptide + Pluronic F-127 | MRSA, Vancomycin-resistant Enterococci, S. epidermidis, CoNS, A. baumannii, P. aeruginosa, K. pneumoniae | More effective in biofilm-killing than the extensively used commercial silver-based antimicrobial product Silver-Sept®. | [118] |
| DspB | - | DspB wound spray combined with Acticoat™, | MRSA, S. epidermidis, A. baumannii, and K. pneumoniae | Combined with treatment with antimicrobial silver wound dressing Acticoat™, the spray decreased viable cell count by 80% compared to a 14% decline with wound dressing alone. | [119] |
| PgaB | GlcN-(β-1,6)-GlcN | Unformulated solution + gentamicin | E. coli, S. carnosus, S. epidermidis, Bordetella pertussis | Potentiate the efficacy of gentamicin in biofilm killing. | [120] |
| PslG | Manp-(β-1,3)- Manp | Unformulated solution and immobilised on material surfaces | P. aeruginosa | Covalently bound PslG significantly reduced P. aeruginosa biofilm formation and surface attachment by ~99.9% (~3-log) compared to untreated surfaces. | [121] |
| Ps1G | - | Immobilization of Ps1G on medical-grade commercial catheter tubing | P. aeruginosa | 3-log and 2-log reduction in bacterial load after 11 and 14 days of enzyme post immobilization respectively. In vivo showed ~a 1.5-log reduction after 24 h) in the colonization of the clinical P. aeruginosa strain. | [122] |
| Ps1G Pe1A | - | Unformulated solutions + Colistin | P. aeruginosa | Effectively inhibited the biofilm formation and significantly disrupted the preformed biofilm with a 58–94% reduction in biofilm biomass. Pe1A also potentiated the colistin efficacy with approx. 50% neutrophil killing. | [123] |
| PelA, Sph3 | (1,4) | Unformulated solutions + amphotericin B, caspofungin, and posaconazole, | A. fumigatus | Effectively disrupted the biofilm with an EC50 of approx. 0.4 nM for both enzymes. PelA and Sph3 also enhanced the antifungal efficacy through increased intracellular penetration. | [124] |
| Alginate lyase Ps1G | Alginate, Manp-(β-1,3)- Manp | Unformulated solutions | Mucoid P. aeruginosa (Clinical isolate) | The comparative study with both glycoside hydrolases showed significant biofilm formation inhibition. | [125] |
| Alginate lyase | Alginate | Unformulated solution + ciprofloxacin and tobramycin | P. aeruginosa | AL enhanced the efficacy of antibiotics through biofilm disruption leading to the significant reduction of biofilm biomass. | [126] |
| Alginate lyase | - | Unformulated solution +Gentamicin and Ceftazidime | P. aeruginosa | The synergy of AL and gentamicin significantly eliminated the mucoid bacteria from biofilm while ceftazidime with AL was more effective against non-mucoid strains. | [127] |
| Alginate lyase | - | AL immobilized on CS nanoparticles in combination with ciprofloxacin | Mucoid P. aeruginosa | Developed nanoparticles significantly inhibited the biofilm formation, and reduced biofilm biomass, density, and thickness in preformed biofilm. | [128] |
| Alginate lyase | - | AL and levofloxacin in high methoxylated pectin microsphere hydrogel | P. aeruginosa | AL enhanced the antimicrobial efficacy of levofloxacin by 35% compared to unformulated solutions. | [129] |
| Alginate lyase | - | AL immobilized on bacterial cellulose membranes + Gentamicin | P. aeruginosa | The combination therapy exhibited a synergistic effect resulting in an 86.5% reduction in viable bacterial cells. | [130] |
| DNase I | eDNA | Unformulated solution + ampicillin, cefotaxime, rifampicin, levofloxacin, and azithromycin | Escherichia coli,Haemophilus influenzae, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pyogenes, and Acinetobacter baumannii | Combined treatment with DNase I enhanced antibiotic efficacy resulting in reduced biofilm biomass and CFU count. | [131] |
| DNase I | - | Unformulated solution + Mg2+ | P. aeruginosa alone or mixed species biofilm with Enterococcus faecalis, Salmonella Typhimurium, and S. aureus | Combined treatment of DNase I with Mg2+ caused 90% biofilm reduction within 5 min against preformed P. aeruginosa biofilms. While this combination was less effective in treating biofilms of mixed species. | [132] |
| DNase I | - | DNase I + Ceftazidime or + ceftazidime linked with chitosan | Burkholderia pseudomallei | DNase I + Ceftazidime caused a 3–4 log reduction in viable cell count in a 2-day-old biofilm. While DNase I + ceftazidime linked with chitosan also significantly inhibited and eradicated pr-formed biofilm. | [133] |
| DNase I | - | DNase-loaded-polylactic-glycolic acid (PLGA) nanoparticles | S. aureus and P. aeruginosa | This combination was effective in preventing biofilm formation and removed >99.8% of the established biofilms. | [134] |
| DNase I | - | Polymer-encapsulated DNase I (n(DNase)) | S. aureus | Effective penetration and log retention time of n (DNase) in biofilm compared to DNase I alone led to 92.2% biofilm disintegration. | [135] |
| DNase I | - | Chitosan nanoparticles (CS NP) loaded with ciprofloxacin and functionalized with DNase I | P. aeruginosa | DNase-functionalized NPs demonstrated significant biofilm formation inhibition and a 2.5-fold reduction in biofilm biomass in preformed biofilm compared to unformulated solutions. | [136] |
| DNase + Proteinase K | eDNA Exoproteins | Unformulated solutions | Multispecies oral biofilms | DNase I significantly inhibited the growth of Fusobacterium nucleatum, Actinomyces oris, Streptococcus oralis Streptococcus mutans, and Candida albicans. Proteinase K caused a significant increase in S. oralis and S. mutans CFUs but reduced the V. dispar and C. albicans CFUs compared to control. CLSM results showed significant biofilm disruption with combined treatment. | [137] |
| α-amylase + Pancreatic protease type-1(PtI) | Exoproteins | Unformulated solutions | S. aureus, MRSA, E. coli | The enzyme combination exhibited significant inhibition of established biofilm (90%, 93%, and 78%), and biofilm prevention (51%, 70%, and 44%) against S. aureus, MRSA, and E. coli respectively. | [138] |
| Serine endo-peptidase protease (Alcalase 2.4 L FG) | - | Oxacillin and penicillin G encapsulated in shellac nanoparticles followed by coating with Alcalase 2.4 L FG | S. aureus | Enhanced the efficacy of antibiotics (~1 × 106 CFU/mL reduction as compared to antibiotic alone) and also exhibited a prompt biofilm degradation. | [139] |
| Alcalase 2.4 L FG | - | Protease functionalized Carbopol nanogels | S. aureus, S. epidermidis, P. aeruginosa, K. pneumoniae, E. faecalis, and E. coli | Nanogels caused a 6-fold reduction in biofilm biomass and a significant decrease in cell density compared to unformulated solutions. Co-treatment of ciprofloxacin and Alcalase coated nanogels produced a 3-log decrease in viable cell count which further led to an undetectable number following co-encapsulation of ciprofloxacin and alcalase in nanogel. | [140] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahsan, A.; Thomas, N.; Barnes, T.J.; Subramaniam, S.; Loh, T.C.; Joyce, P.; Prestidge, C.A. Lipid Nanocarriers-Enabled Delivery of Antibiotics and Antimicrobial Adjuvants to Overcome Bacterial Biofilms. Pharmaceutics 2024, 16, 396. https://doi.org/10.3390/pharmaceutics16030396
Ahsan A, Thomas N, Barnes TJ, Subramaniam S, Loh TC, Joyce P, Prestidge CA. Lipid Nanocarriers-Enabled Delivery of Antibiotics and Antimicrobial Adjuvants to Overcome Bacterial Biofilms. Pharmaceutics. 2024; 16(3):396. https://doi.org/10.3390/pharmaceutics16030396
Chicago/Turabian StyleAhsan, Anam, Nicky Thomas, Timothy J. Barnes, Santhni Subramaniam, Thou Chen Loh, Paul Joyce, and Clive A. Prestidge. 2024. "Lipid Nanocarriers-Enabled Delivery of Antibiotics and Antimicrobial Adjuvants to Overcome Bacterial Biofilms" Pharmaceutics 16, no. 3: 396. https://doi.org/10.3390/pharmaceutics16030396