
The Development of a Stable Peptide-Loaded Long-Acting Injection Formulation through a Comprehensive Understanding of Peptide Degradation Mechanisms: A QbD-Based Approach

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment and Instruments
2.3. Degradation Mechanism Studies of ACTY116
2.3.1. Method for ACTY116 Impurity Analysis
- Chromatographic column: XSelect CSH C18, 150 × 4.6 mm, 5 μm;
- Wavelength: 210 nm;
- Column temperature: 40 °C;
- Flow rate: 1 mL/min;
- Injection volume: 25 μL;
- Mobile phase:
- ○
- A: Using phosphate buffer pH 6.5 (containing 20 mM sodium perchlorate);
- ○
- B: Using acetonitrile–water (v/v) = 80:20.
2.3.2. Forced Degradation Studies
2.4. LAI Formulation Design
2.4.1. Preparation of Microspheres
2.4.2. Preparation of Phospholipid-Based In Situ Gel
2.4.3. Preparation of PLGA-Based In Situ Gel
2.4.4. Characterization of Different Formulations
2.5. Identification of CQAs
2.5.1. Impurity Profiles (CQA1) for Different Formulations
2.5.2. Other Critical Quality Attributes (CQA2) for Different Formulations
2.6. Risk Assessment and Control Strategies
2.7. Updated CQAs for Different Formulations
2.8. Pharmacokinetic Study in Rats
2.8.1. LC-MS/MS Method
2.8.2. PK Study in Rats
2.9. Evaluation of Target Product Profile Achievement
3. Results
3.1. Quality Target Product Profile
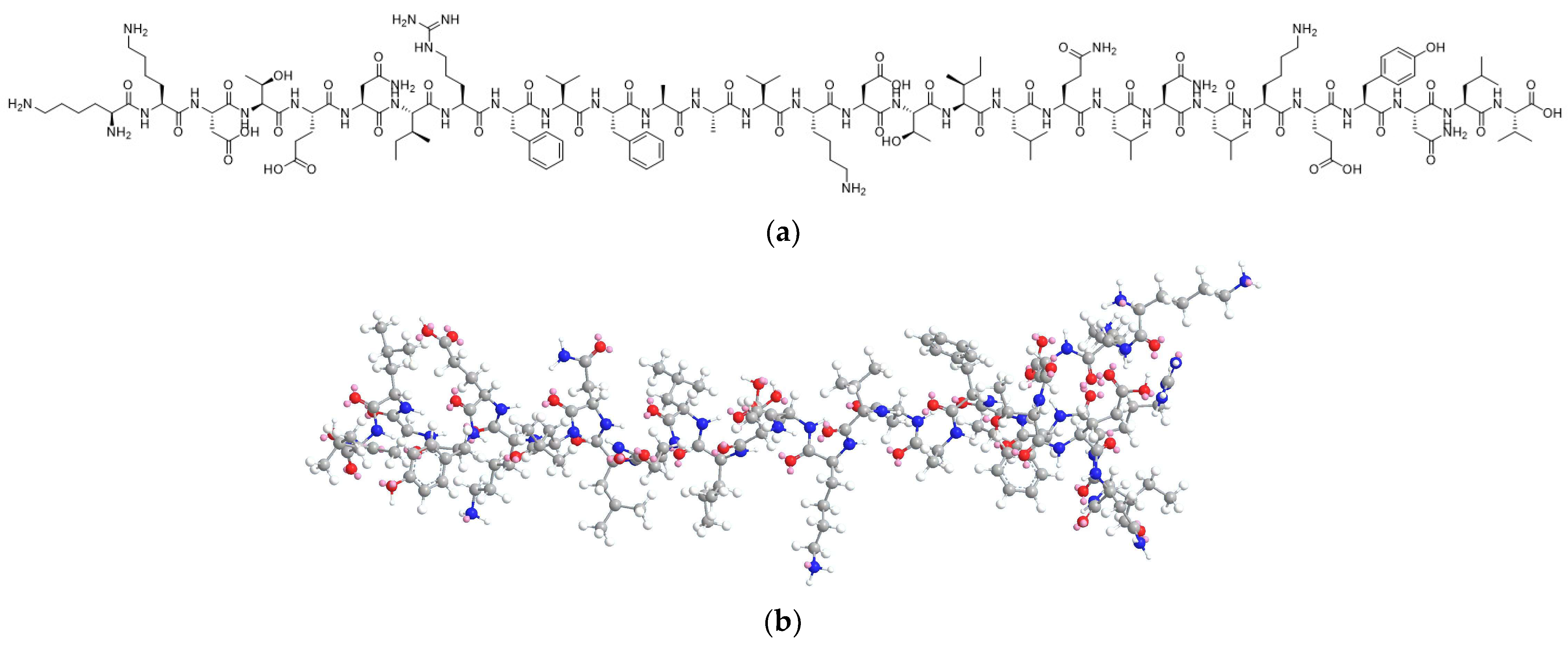
3.2. Degradation Mechanism Studies on ACTY116
3.3. Characterization of Different LAI Formulations
3.4. Initial CQAs
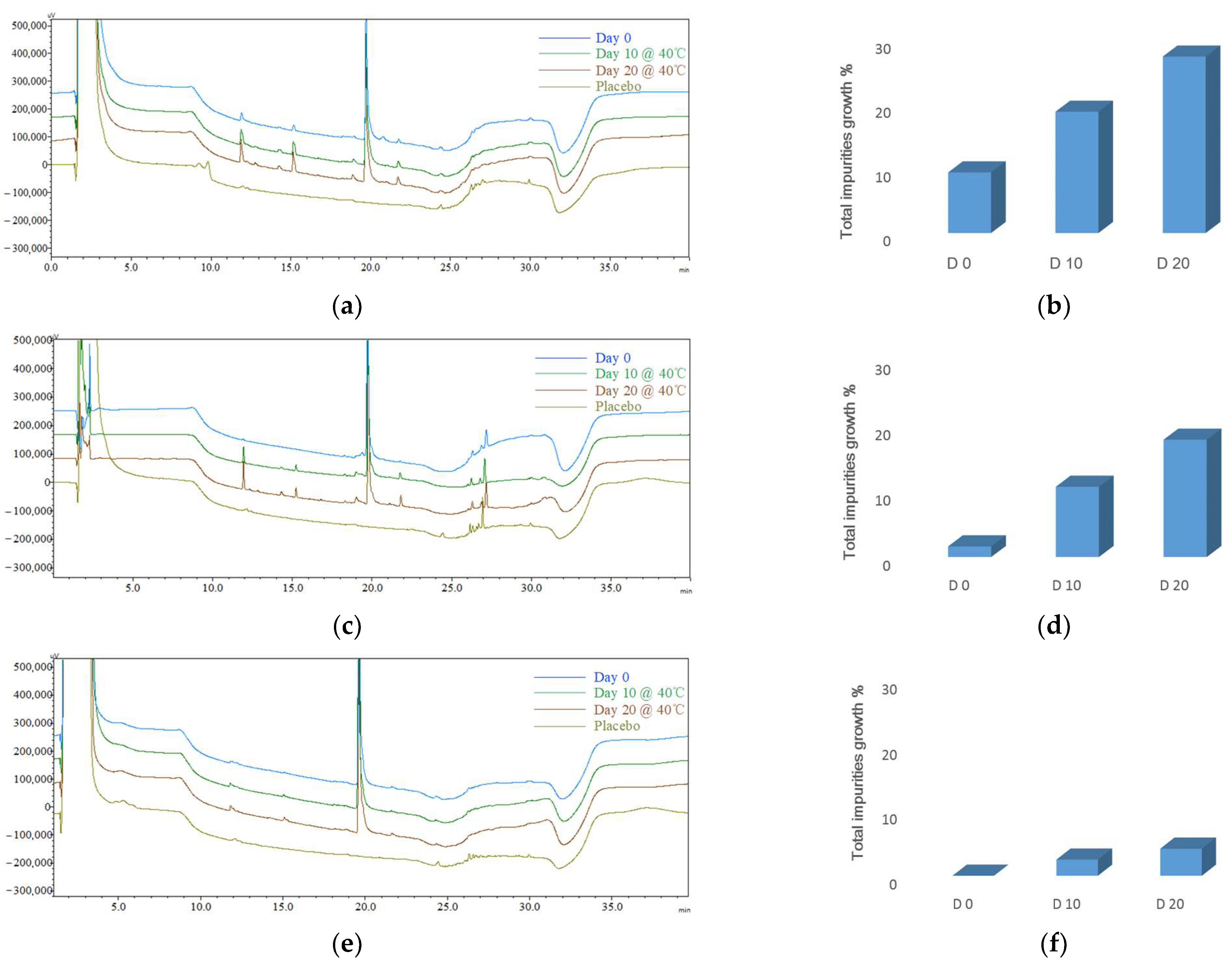
3.4.1. Initial Impurity Profiles (CQA1) for Different Formulations
3.4.2. Initial CQA2 for Different Formulations
3.5. Risk Assessment and Control Strategies
3.5.1. Critical Material Attributes (CMAs) for Excipients
3.5.2. CPP Control Strategies
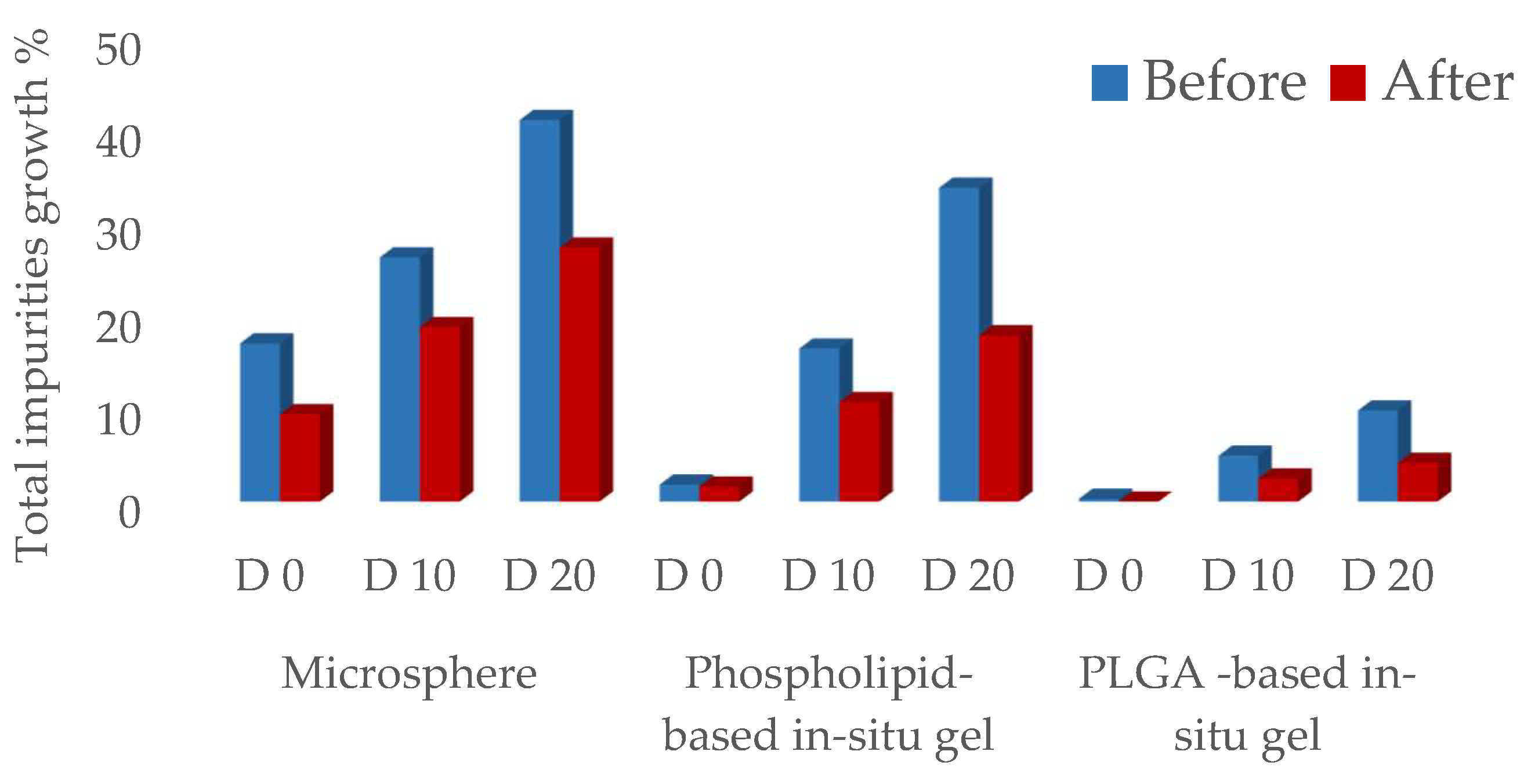
3.6. Updated CQAs for Different Formulations
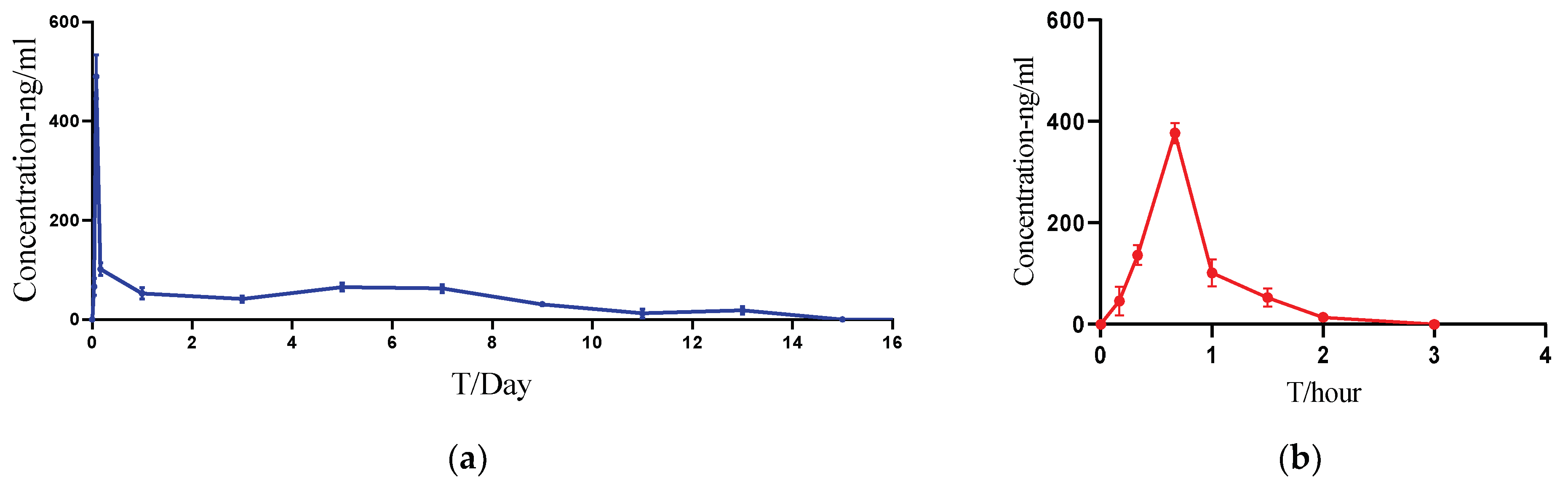
3.7. Pharmacokinetic Studies in Rats
3.8. Evaluation of Target Product Profile Achievement
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Liu, Y.; Lu, X.L.; Li, X.H.; Zhang, H.G. Transmembrane transport of the gαq protein carboxyl terminus imitation polypeptide gcip-27. Eur. J. Pharm. Sci. 2013, 49, 791–799. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, X.L.; Yang, H.; Li, X.H.; Zhang, H.G. Effects of polypeptide drug huilixinkang on cardiac hypertrophy and expressions of myosin heavy chain in mice. Chin. Pharm. J. 2011, 46, 1566–1569. [Google Scholar]
- Lu, X.L.; Tong, Y.F.; Liu, Y.; Xu, Y.L.; Yang, H.; Zhang, G.Y.; Li, X.H.; Zhang, H.G. Gαq protein carboxyl terminus imitation polypeptide gcip-27 improves cardiac function in chronic heart failure rats. PLoS ONE 2015, 10, e0121007. [Google Scholar]
- Blessy, M.R.D.P.; Patel, R.D.; Prajapati, P.N.; Agrawal, Y.K. Development of forced degradation and stability indicating studies of drugs—A review. J. Pharm. Anal. 2014, 4, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Naazneen, S.; Sridevi, A. Development of assay method and forced degradation study of valsartan and sacubitril by RP-HPLC in tablet formulation. Int. J. Appl. Pharm. 2017, 9, 9–15. [Google Scholar] [CrossRef]
- Jimmidi, R. Synthesis and applications of peptides and peptidomimetics in drug discovery. Eur. J. Org. Chem. 2023, 26, e202300028. [Google Scholar] [CrossRef]
- Nugrahadi, P.P.; Hinrichs, W.L.; Frijlink, H.W.; Schöneich, C.; Avanti, C. Designing formulation strategies for enhanced stability of therapeutic peptides in aqueous solutions: A review. Pharmaceutics 2023, 15, 935. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lu, A.; Wang, X.; Belahadj, Z.; Wang, J.; Zhang, Q. A review of existing strategies for designing long-acting parenteral formulations: Focus on underlying mechanisms, and future perspectives. Acta Pharm. Sin. B. 2021, 11, 2396–2415. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.R.; Kulkarni, V.V.; Pokhrel, S.; Akram, H.; Abdelgadir, A.; Chatterjee, A.; Khan, S. Comparing the efficacy and safety of obeticholic acid and semaglutide in patients with non-alcoholic fatty liver disease: A systematic review. Cureus 2022, 14. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The discovery and development of liraglutide and semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Citrome, L. Paliperidone palmitate—Review of the efficacy, safety and cost of a new second-generation depot antipsychotic medication. Int. J. Clin. Pract. 2010, 64, 216–239. [Google Scholar] [CrossRef]
- Li, G.; Rui-Guo, Z.; Yu-Ting, Q.; Yun-Chun, C.; Hua-Ning, W.; Psychosomatic, D.O. Efficacy and Safety of Paliperidone Palmitate or Long-acting Injectable Risperidone in Patients with Schizophrenia. Prog. Mod. Biomed. 2017, 17, 688–691. [Google Scholar]
- Fowler, J.E., Jr.; Flanagan, M.; Gleason, D.M.; Klimberg, I.W.; Gottesman, J.E.; Sharifi, R. Evaluation of an implant that delivers leuprolide for 1 year for the palliative treatment of prostate cancer. Urology 2000, 55, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y. Injectable, long-acting plga formulations: Analyzing plga and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Asadujjaman, M.; Jee, J.P. Long acting injectable formulations: The state of the arts and challenges of poly (lactic-co-glycolic acid) microsphere, hydrogel, organogel and liquid crystal. J. Pharm. Investig. 2019, 49, 459–476. [Google Scholar] [CrossRef]
- O’Brien, M.N.; Jiang, W.; Wang, Y.; Loffredo, D.M. Challenges and opportunities in the development of complex generic long-acting injectable drug products. J. Control. Release 2021, 336, 144–158. [Google Scholar] [CrossRef]
- Zhou, J.; Walker, J.; Ackermann, R.; Olsen, K.; Hong, J.K.; Wang, Y.; Schwendeman, S.P. Effect of manufacturing variables and raw materials on the composition-equivalent plga microspheres for 1-month controlled release of leuprolide. Mol. Pharm. 2020, 17, 1502–1515. [Google Scholar] [CrossRef]
- Park, K.; Otte, A.; Sharifi, F.; Garner, J.; Skidmore, S.; Park, H.; Jhon, Y.K.; Qin, B.; Wang, Y. Formulation composition, manufacturing process, and characterization of poly (lactide-co-glycolide) microparticles. J. Control. Release 2021, 329, 1150–1161. [Google Scholar] [CrossRef]
- Nair, H.A.; Begum, N. Development and evaluation of a poloxamer- and chitosan-based in situ gel-forming injectable depot. Asian J. Pharm. Clin. Res. 2020, 13, 36–41. [Google Scholar] [CrossRef]
- Muddineti, O.S.; Omri, A. Current trends in plga based long-acting injectable products: The industry perspective. Expert Opin. Drug Deliv. 2022, 19, 559–576. [Google Scholar] [CrossRef]
- Somayaji, M.R.; Das, D.; Przekwas, A. A new level a type ivivc for the rational design of clinical trials toward regulatory approval of generic polymeric long-acting injectables. Clin. Pharmacokinet. 2016, 55, 1179–1190. [Google Scholar] [CrossRef]
- Butreddy, A.; Gaddam, R.P.; Kommineni, N.; Dudhipala, N.; Voshavar, C. Plga/pla-based long-acting injectable depot microspheres in clinical use: Production and characterization overview for protein/peptide delivery. Int. J. Mol. Sci. 2021, 22, 8884. [Google Scholar] [CrossRef]
- Silva, A.T.C.R.; Cardoso, B.C.O.; e Silva, M.E.S.R.; Freitas, R.F.S.; Sousa, R.G. Synthesis, characterization, and study of plga copolymer in vitro degradation. J. Biomater. Nanobiotechnol. 2015, 6, 8. [Google Scholar] [CrossRef]
- Mir, M.; Ahmed, N.; ur Rehman, A. Recent applications of plga based nanostructures in drug delivery. Colloids Surf. B Biointerfaces 2017, 159, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Tan, W.S.; Ho, K.L.; Mariatulqabtiah, A.R.; Abu Kasim, N.H.; Abd Rahman, N.; Wong, T.W.; Chee, C.F. Challenges and Complications of Poly(lactic-co-glycolic acid)-Based Long-Acting Drug Product Development. Pharmaceutics 2022, 14, 614. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, Y.; Xiang, N.; Zhong, Y.; Gong, T.; Zhang, Z.R.; Fu, Y. Long-Acting Phospholipid Gel of Exenatide for Long-Term Therapy of Type II Diabetes. Pharm. Res. 2016, 33, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Nanxi, X.; Yu, Z.; Xinyi, H.E.; Jinjie, Z.; Xun, S.; Tao, G. Preparation and pharmacokinetic study of Huperzine A phospholipid in situ-gel. West China J. Pharm. Sci. 2017, 32, 136–146. [Google Scholar]
- Li, H.; Liu, T.; Zhu, Y.; Fu, Q.; Wu, W.; Deng, J.; Shi, S. An in situ-forming phospholipid-based phase transition gel prolongs the duration of local anesthesia for ropivacaine with minimal toxicity. Acta Biomater. 2017, 58, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Dai, Z.; Zhang, Z.; Kou, X.; You, X.; Sun, H.; Zhu, H. Fabrication of oral nanovesicle in-situ gel based on Epigallocatechin gallate phospholipid complex: Application in dental anti-caries. Eur. J. Pharmacol. 2021, 897, 173951. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Devi, M.S.; Babu, G.R.; Mulukuri, N.S. A new stability indicating RP-HPLC method for the simultaneous estimation of Diloxanide and Ornidazole in bulk and Pharmaceutical Dosage forms. Res. J. Pharm. Technol. 2017, 12, 4247–4254. [Google Scholar] [CrossRef]
- Lakka NSKuppan, C.; Srinivas, K.S.; Yarra, R. Separation and Characterization of New Forced Degradation Products of Dasatinib in Tablet Dosage Formulation Using LC-MS and Stability-Indicating HPLC Methods. Chromatographia 2020, 83, 947–962. [Google Scholar] [CrossRef]
- Rathor, S.; Sherje, A. Forced degradation studies of tizanidine hydrochloride and development of validated stability-indicating RP-HPLC method. Indian Drugs 2021, 58, 50–55. [Google Scholar] [CrossRef]
- Li, Z.; Mu, H.; Larsen, S.W.; Jensen, H.; Østergaard, J. Initial leuprolide acetate release from poly (d, l-lactide-co-glycolide) in situ forming implants as studied by ultraviolet–visible imaging. Mol. Pharm. 2020, 17, 4522–4532. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; El-Megrab, N.A.; El-Nahas, H.M. Optimization of injectable plga in-situ forming implants of anti-psychotic risperidone via box-behnken design. J. Drug Deliv. Sci. Technol. 2020, 58, 101803. [Google Scholar] [CrossRef]
- Shukr, M.H.; Ismail, S.; El-Hossary, G.G.; El-Shazly, A.H. Design and evaluation of mucoadhesive in situ liposomal gel for sustained ocular delivery of travoprost using two steps factorial design. J. Drug Deliv. Sci. Technol. 2021, 61, 102333. [Google Scholar] [CrossRef]
- European Pharmacopoeia 11.0; European Directorate for the Quality of Medicines & Healthcare: Strasbourg, France, 2022.
- Doleschall, F.; KemÉNy, Z.; Recseg, K.; Kővári, K. A new analytical method to monitor lipid peroxidation during bleaching. Eur. J. Lipid Sci. Technol. 2002, 104, 14–18. [Google Scholar] [CrossRef]
- Lai-Lin, Z.; Qing-He, Z.; Jie-Sheng, Z.; Shu-Li, W. Effects of storage conditions on oil-making quality of sesame seeds. J. Henan Univ. Technol. 2015, 36, 47–51. [Google Scholar]
- Vigani, B.; Rossi, S.; Sandri, G.; Bonferoni, M.C.; Caramella, C.M.; Ferrari, F. Recent advances in the development of in situ gelling drug delivery systems for non-parenteral administration routes. Pharmaceutics 2020, 12, 859. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; El-Megrab, N.A.; El-Nahas, H.M. An overview of plga in-situ forming implants based on solvent exchange technique: Effect of formulation components and characterization. Pharm. Dev. Technol. 2021, 26, 709–728. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Park, I.S.; Kim, S.; Yim, Y.; Park, G.; Choi, J.; Won, C.; Min, D.H. Multifunctional synthetic nano-chaperone for peptide folding and intracellular delivery. Nat. Commun. 2022, 13, 4568. [Google Scholar] [CrossRef] [PubMed]
- Massaro, M.; Licandro, E.; Cauteruccio, S.; Lazzara, G.; Liotta, L.F.; Notarbartolo, M.; Raymo, F.M.; Sánchez-Espejo, R.; Viseras-Iborra, C.; Riela, S. Nanocarrier based on halloysite and fluorescent probe for intracellular delivery of peptide nucleic acids. J. Colloid Interface Sci. 2022, 620, 221–233. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time/Min | 0 | 5 | 6 | 20 | 23 | 28 | 30 | 40 |
|---|---|---|---|---|---|---|---|---|
| A% | 90 | 90 | 75 | 50 | 10 | 10 | 90 | 90 |
| B% | 10 | 10 | 25 | 50 | 90 | 90 | 10 | 10 |
| Microsphere | Phospholipid-Based In Situ Gel | PLGA-Based In Situ Gel | |
|---|---|---|---|
| Ingredients | ACTY116 PLGA DCM PVA Mannitol | ACTY116 Soybean phospholipid MCT | ACTY116 PLGA NMP |
| Temperature/°C | −40 | −40 | −20 | −20 | 0 | 25 | 25 |
|---|---|---|---|---|---|---|---|
| Duration/min | 30 | 180 | 120 | 270 | 180 | 120 | 600 |
| Pressure/Pa | — | — | 10–16 | 10–16 | 10–16 | 10–16 | 10–16 |
| Degradation Condition | Control Sample | Thermal Stress | Photolytic Stress | Oxidative Stress | Hydrolytic Stress | Acidic Hydrolytic Stress |
|---|---|---|---|---|---|---|
| Total impurities % | 2.2 | 2.3 | 2.2 | 10.1 | 9.2 | 34.1 |
| Particle Size (μm) | EE% | Viscosity (cP) | ||||
|---|---|---|---|---|---|---|
| D(10) | D(50) | D(90) | D(4,3) | |||
| F1 | 5.12 | 16.6 | 38.3 | 21.9 | 29.8 | NA |
| F2 | 6.48 | 26.6 | 104 | 41.6 | NA | 142 |
| F3 | 7.11 | 28.5 | 107 | 44.1 | NA | 115 |
| F1 | F2 | F3 | |
|---|---|---|---|
| Day 0 | 17.09 ± 1.18% | 1.83 ± 0.87% | 0.31 ± 0.08% |
| Day 10 | 26.39 ± 4.84% | 16.54 ± 0.33% | 4.95 ± 0.69% |
| Day 20 | 41.22 ± 0.48% | 33.90 ± 3.52% | 9.85 ± 2.74% |
| Water% | IA | II | IP | AV | |
|---|---|---|---|---|---|
| Microspheres (F1) | 2.37 | 0 | 0 | 0.2 | 0 |
| Phospholipid-based in situ gel (F2) | 0.6 | 2.9 | 37.2 | 2.6 | 2.1 |
| PLGA-based in situ gel (F3) | 0.03 | 0 | 0 | 0 | 0 |
| Water% | IA | II | IP | AV | ||
|---|---|---|---|---|---|---|
| F1 | PLGA | 0.01 | 0 | 0 | 0 | 0 |
| DCM | 0.06 | 0 | 0 | 0 | 0 | |
| F2 | Soybean phospholipid | 1.4 | 8.3 | 105.3 | 2.8 | 0.2 |
| MCT | 0.1 | 0.2 | 0.9 | 1.4 | 0.6 | |
| F3 | PLGA | 0.01 | 0 | 0 | 0 | 0 |
| NMP | 0.03 | 0 | 0 | 0 | 0 |
| Water% | IA | II | IP | AV | ||
|---|---|---|---|---|---|---|
| Soybean phospholipid | F2 | 1.4 | 8.3 | 105.3 | 2.8 | 0.2 |
| F5 | 1.1 | 1.6 | 93.7 | 1.3 | 0.1 | |
| MCT | F2 | 0.1 | 0.2 | 0.9 | 1.4 | 0.6 |
| F5 | 0.1 | 0.1 | 0.8 | 0.3 | 0.2 |
| Microsphere F4 | Phospholipid-Based In Situ Gel F5 | PLGA-Based In Situ Gel F6 | ||
|---|---|---|---|---|
| CQA1 (impurity profiles) | Day 0 | 9.42 ± 1.31% | 1.62 ± 0.79% | 0.00% |
| Day 10 | 18.85 ± 1.06% | 10.75 ± 0.81% | 2.48 ± 0.42% | |
| Day 20 | 27.43 ± 3.33% | 17.95 ± 1.67% | 4.17 ± 0.42% | |
| CQA2 | Water% | 1.45 | 0.4 | 0.01 |
| IA | 0 | 0.6 | 0 | |
| II | 0 | 35.5 | 0 | |
| IP | 0 | 0.7 | 0 | |
| AV | 0 | 0.6 | 0 | |
| Control strategies | CMAs | - | Selection of excipients with superior CMAs | - |
| CPPs |
| Oxygen in headspace ≤ 0.1% | Oxygen in headspace ≤ 0.1% |
| PK Parameters | Cmax (ng/mL) | AUClast (h·ng/mL) | Tmax (h) | HL_Lambda_z (h) | MRTlast (h) | Tlast (h) |
|---|---|---|---|---|---|---|
| ACTY116 Solution | 377.4 ± 20.3 | 240.5 ± 29.1 | 0.67 | 0.351 ± 0.021 | 0.796 ± 0.024 | 2 ± 0 |
| ACTY116 LAI formulation | 489.8 ± 44.3 | 14,385.6 ± 1063.2 | 1.99 | 100.4 ± 50.4 | 117.5 ± 7.2 | 312 ± 19.2 |
| The Total Impurities’ Growth | |||
|---|---|---|---|
| During Preparation | 10 Days @ 40 °C | 20 Days @ 40 °C | |
| Target product impurity profile | ≤1.0% | ≤3.5% | ≤7.0% |
| Microspheres (F4) | 9.42 ± 1.31% | 18.85 ± 1.06% | 27.43 ± 3.33% |
| Phospholipid-based in situ gel (F5) | 1.62 ± 0.79% | 10.75 ± 0.81% | 17.95 ± 1.67% |
| PLGA-based in situ gel (F6) | 0 | 2.48 ± 0.42% | 4.17 ± 0.42% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; Wang, J.; Zhou, X.; Li, X. The Development of a Stable Peptide-Loaded Long-Acting Injection Formulation through a Comprehensive Understanding of Peptide Degradation Mechanisms: A QbD-Based Approach. Pharmaceutics 2024, 16, 266. https://doi.org/10.3390/pharmaceutics16020266
Xiong Y, Wang J, Zhou X, Li X. The Development of a Stable Peptide-Loaded Long-Acting Injection Formulation through a Comprehensive Understanding of Peptide Degradation Mechanisms: A QbD-Based Approach. Pharmaceutics. 2024; 16(2):266. https://doi.org/10.3390/pharmaceutics16020266
Chicago/Turabian StyleXiong, Yingxin, Jiawei Wang, Xing Zhou, and Xiaohui Li. 2024. "The Development of a Stable Peptide-Loaded Long-Acting Injection Formulation through a Comprehensive Understanding of Peptide Degradation Mechanisms: A QbD-Based Approach" Pharmaceutics 16, no. 2: 266. https://doi.org/10.3390/pharmaceutics16020266