
Update and Application of a Deep Learning Model for the Prediction of Interactions between Drugs Used by Patients with Multiple Sclerosis

Abstract
:1. Background
2. Methods
2.1. Patient Cohort
2.2. Drug Data and Gold Standard DDI Dataset
2.3. Food Content Data
2.4. DeepDDI Model Update and Performance Evaluation
2.5. Prediction of DDIs and DFIs
2.6. Post-Processing of DDI Results
2.7. DFI Network Visualization
3. Results
3.1. Drugs Used by the Patients with MS
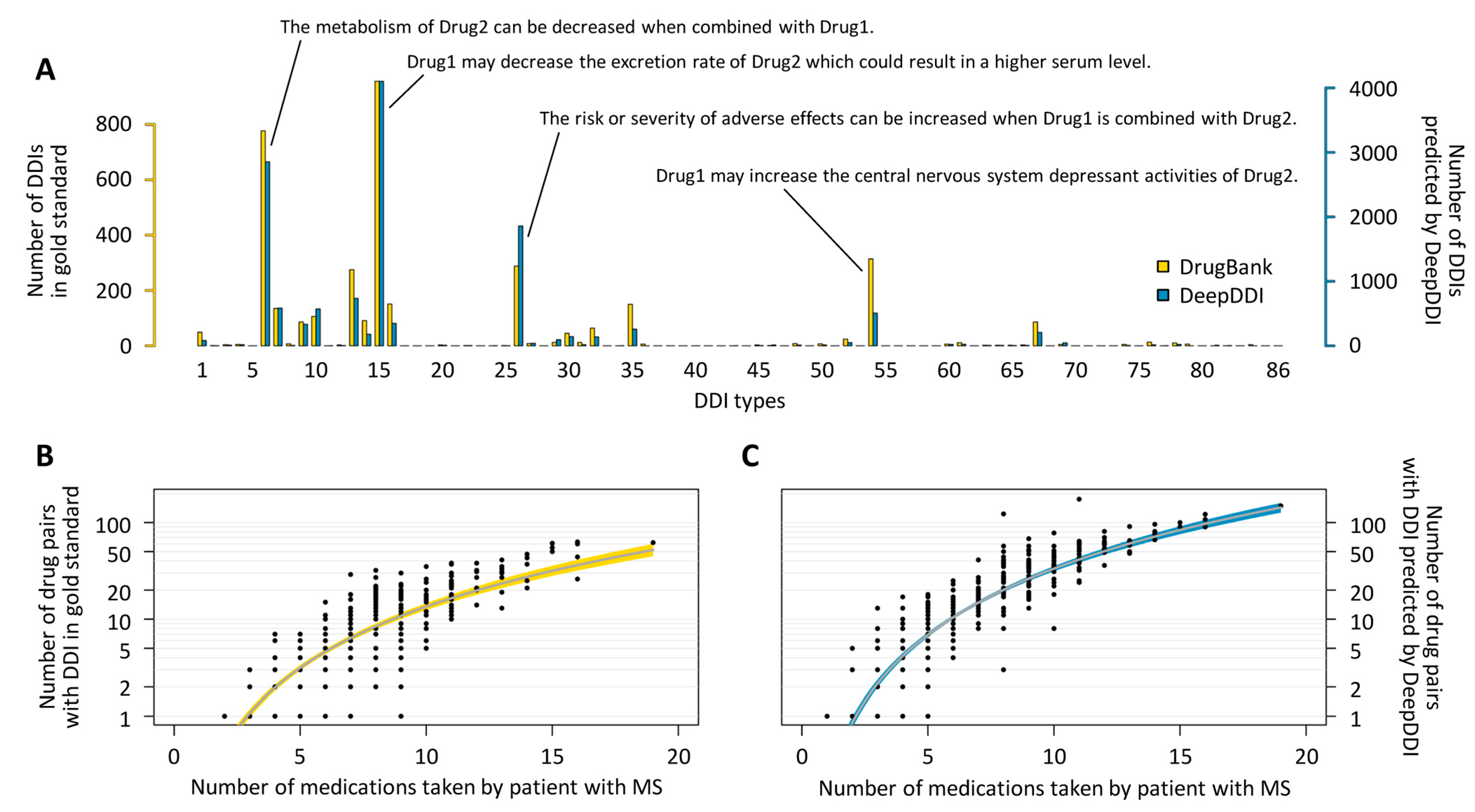
3.2. Update and Application of the DeepDDI Model
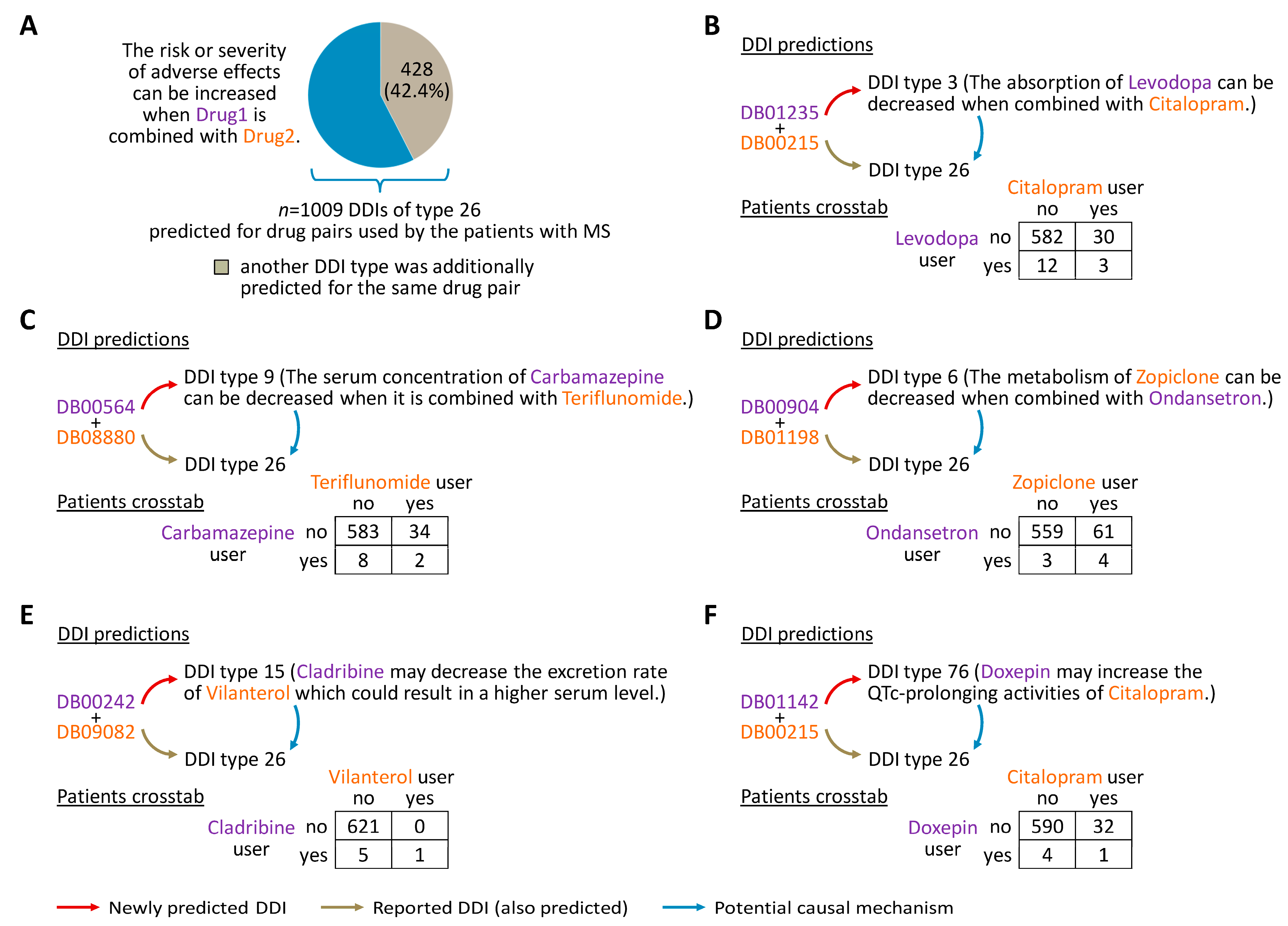
3.3. DDIs of Potential Relevance in the Patients
3.4. Mechanisms That May Explain Adverse Drug Effects
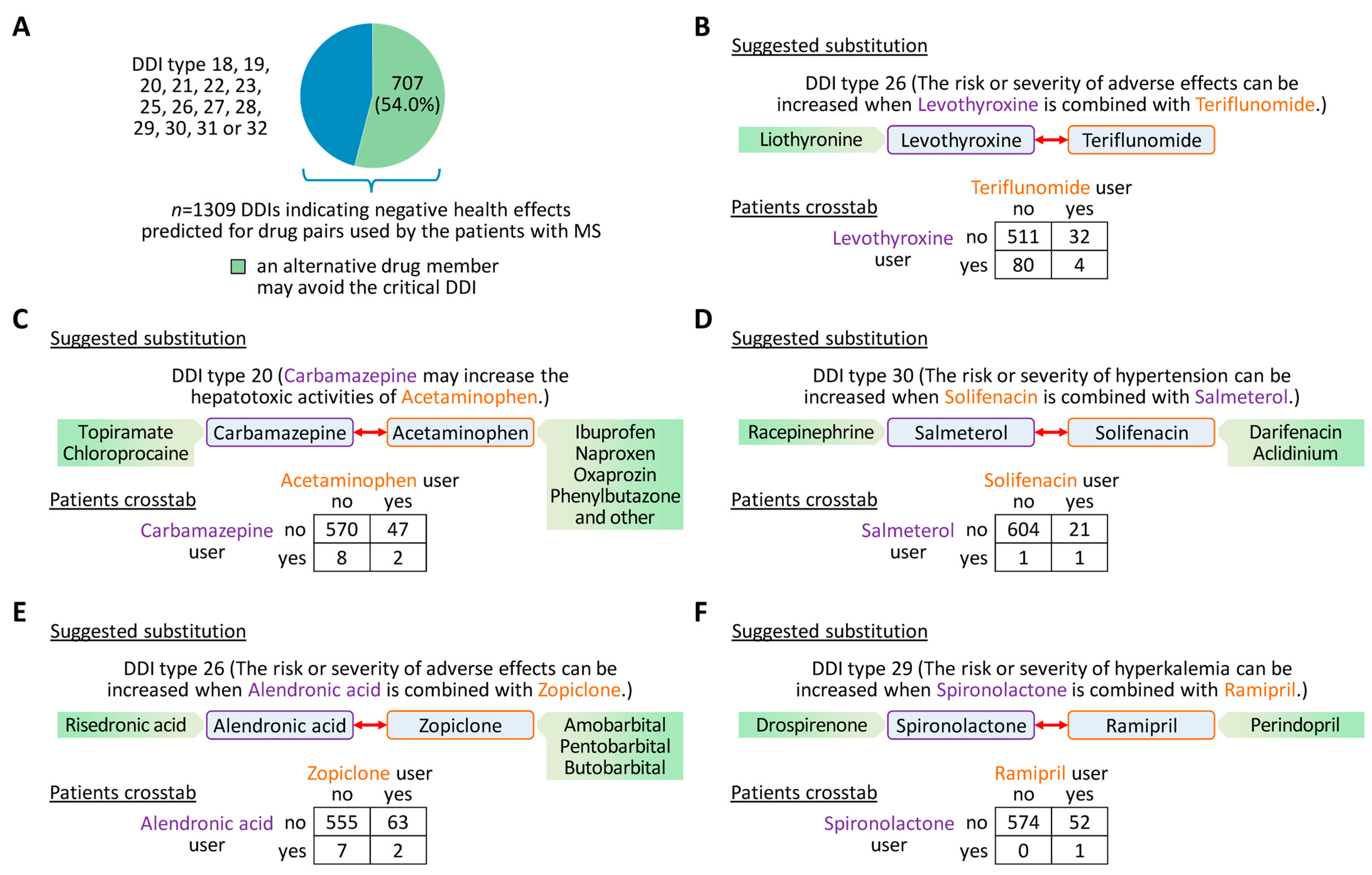
3.5. Suggestions for Drug Substitution to Avoid Critical DDIs
3.6. DFIs That May Alter Drug Concentrations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; van der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. 2020, 26, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Eichstädt, K.; Ellenberger, D.; Flachenecker, P.; Friede, T.; Haas, J.; Kleinschnitz, C.; Pöhlau, D.; Rienhoff, O.; Stahmann, A.; et al. Symptomatology and symptomatic treatment in multiple sclerosis: Results from a nationwide MS registry. Mult. Scler. 2019, 25, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Goodin, D.S. The epidemiology of multiple sclerosis: Insights to a causal cascade. Handb. Clin. Neurol. 2016, 138, 173–206. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cree, B.A.C. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390.e2. [Google Scholar] [CrossRef]
- Yang, J.H.; Rempe, T.; Whitmire, N.; Dunn-Pirio, A.; Graves, J.S. Therapeutic Advances in Multiple Sclerosis. Front. Neurol. 2022, 13, 824926. [Google Scholar] [CrossRef]
- Wiendl, H.; Gold, R.; Berger, T.; Derfuss, T.; Linker, R.; Mäurer, M.; Aktas, O.; Baum, K.; Berghoff, M.; Bittner, S.; et al. Multiple Sclerosis Therapy Consensus Group (MSTCG): Position statement on disease-modifying therapies for multiple sclerosis (white paper). Ther. Adv. Neurol. Disord. 2021, 14, 17562864211039648. [Google Scholar] [CrossRef]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef] [PubMed]
- Moiola, L.; Rommer, P.S.; Zettl, U.K. Prevention and management of adverse effects of disease modifying treatments in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Rommer, P.S.; Zettl, U.K. Managing the side effects of multiple sclerosis therapy: Pharmacotherapy options for patients. Expert. Opin. Pharmacother. 2018, 19, 483–498. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736. [Google Scholar] [CrossRef]
- Carnero Contentti, E.; Correale, J. Current Perspectives: Evidence to Date on BTK Inhibitors in the Management of Multiple Sclerosis. Drug Des. Dev. Ther. 2022, 16, 3473–3490. [Google Scholar] [CrossRef]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Hartung, H.P.; Barnett, M. New drugs for multiple sclerosis: New treatment algorithms. Curr. Opin. Neurol. 2022, 35, 262–270. [Google Scholar] [CrossRef]
- Kochs, L.; Wegener, S.; Sühnel, A.; Voigt, K.; Zettl, U.K. The use of complementary and alternative medicine in patients with multiple sclerosis: A longitudinal study. Complement. Ther. Med. 2014, 22, 166–172. [Google Scholar] [CrossRef]
- Rommer, P.S.; König, N.; Sühnel, A.; Zettl, U.K. Coping behavior in multiple sclerosis-complementary and alternative medicine: A cross-sectional study. CNS Neurosci. Ther. 2018, 24, 784–789. [Google Scholar] [CrossRef]
- Frahm, N.; Hecker, M.; Zettl, U.K. Polypharmacy in Chronic Neurological Diseases: Multiple Sclerosis, Dementia and Parkinson’s Disease. Curr. Pharm. Des. 2021, 27, 4008–4016. [Google Scholar] [CrossRef]
- Hecker, M.; Frahm, N.; Bachmann, P.; Debus, J.L.; Haker, M.C.; Mashhadiakbar, P.; Langhorst, S.E.; Baldt, J.; Streckenbach, B.; Heidler, F.; et al. Screening for severe drug-drug interactions in patients with multiple sclerosis: A comparison of three drug interaction databases. Front. Pharmacol. 2022, 13, 946351. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and induction of CYP enzymes in humans: An update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic Drug-Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Spanakis, M.; Patelarou, E.; Patelarou, A. Drug-Food Interactions with a Focus on Mediterranean Diet. Appl. Sci. 2022, 12, 10207. [Google Scholar] [CrossRef]
- Jalusic, K.O.; Ellenberger, D.; Rommer, P.; Stahmann, A.; Zettl, U.; Berger, K. Effect of applying inclusion and exclusion criteria of phase III clinical trials to multiple sclerosis patients in routine clinical care. Mult. Scler. 2021, 27, 1852–1863. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine Learning for Integrating Data in Biology and Medicine: Principles, Practice, and Opportunities. Inf. Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef]
- Lu, C.; Di, L. In vitro and in vivo methods to assess pharmacokinetic drug-drug interactions in drug discovery and development. Biopharm. Drug Dispos. 2020, 41, 3–31. [Google Scholar] [CrossRef]
- Vinarov, Z.; Butler, J.; Kesisoglou, F.; Koziolek, M.; Augustijns, P. Assessment of food effects during clinical development. Int. J. Pharm. 2023, 635, 122758. [Google Scholar] [CrossRef]
- Roblek, T.; Vaupotic, T.; Mrhar, A.; Lainscak, M. Drug-drug interaction software in clinical practice: A systematic review. Eur. J. Clin. Pharmacol. 2015, 71, 131–142. [Google Scholar] [CrossRef]
- Marcath, L.A.; Xi, J.; Hoylman, E.K.; Kidwell, K.M.; Kraft, S.L.; Hertz, D.L. Comparison of Nine Tools for Screening Drug-Drug Interactions of Oral Oncolytics. J. Oncol. Pract. 2018, 14, e368–e374. [Google Scholar] [CrossRef]
- Hines, L.E.; Malone, D.C.; Murphy, J.E. Recommendations for generating, evaluating, and implementing drug-drug interaction evidence. Pharmacotherapy 2012, 32, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Kheshti, R.; Aalipour, M.; Namazi, S. A comparison of five common drug-drug interaction software programs regarding accuracy and comprehensiveness. J. Res. Pharm. Pract. 2016, 5, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sancar, M.; Kaşik, A.; Okuyan, B.; Batuhan, S.; Izzettin, F.V. Determination of Potential Drug-Drug Interactions Using Various Software Programs in a Community Pharmacy Setting. Turk. J. Pharm. Sci. 2019, 16, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Armahizer, M.J.; Kane-Gill, S.L.; Smithburger, P.L.; Anthes, A.M.; Seybert, A.L. Comparing drug-drug interaction severity ratings between bedside clinicians and proprietary databases. Int. Sch. Res. Not. 2013, 2013, 347346. [Google Scholar] [CrossRef]
- Zheng, W.Y.; Richardson, L.C.; Li, L.; Day, R.O.; Westbrook, J.I.; Baysari, M.T. Drug-drug interactions and their harmful effects in hospitalised patients: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2018, 74, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Cao, P.; Wang, Y.; Xie, F.; Ma, J.; Yu, M.; Wang, J.; Xu, Y.; Zhang, Y.; Wan, J. A Review of Approaches for Predicting Drug-Drug Interactions Based on Machine Learning. Front. Pharmacol. 2022, 12, 814858. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Jeon, J.; Kim, H.U. Recent development of machine learning models for the prediction of drug-drug interactions. Korean J. Chem. Eng. 2023, 40, 276–285. [Google Scholar] [CrossRef]
- Lin, X.; Dai, L.; Zhou, Y.; Yu, Z.G.; Zhang, W.; Shi, J.Y.; Cao, D.S.; Zeng, L.; Chen, H.; Song, B.; et al. Comprehensive evaluation of deep and graph learning on drug-drug interactions prediction. Brief. Bioinform. 2023, 24, bbad235. [Google Scholar] [CrossRef]
- Percha, B.; Altman, R.B. Informatics confronts drug-drug interactions. Trends Pharmacol. Sci. 2013, 34, 178–184. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, Z. Machine learning-based prediction of drug-drug interactions by integrating drug phenotypic, therapeutic, chemical, and genomic properties. J. Am. Med. Inform. Assoc. 2014, 21, e278–e286. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci. USA 2018, 115, E4304–E4311. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, X.; Qiu, Y.; Xia, J.; Zhang, W.; Liu, S. A multimodal deep learning framework for predicting drug-drug interaction events. Bioinformatics 2020, 36, 4316–4322. [Google Scholar] [CrossRef] [PubMed]
- Udrescu, M.; Ardelean, S.M.; Udrescu, L. The curse and blessing of abundance-the evolution of drug interaction databases and their impact on drug network analysis. Gigascience 2022, 12, giad011. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Debus, J.L.; Bachmann, P.; Frahm, N.; Mashhadiakbar, P.; Langhorst, S.E.; Streckenbach, B.; Baldt, J.; Heidler, F.; Hecker, M.; Zettl, U.K. Associated factors of potential drug-drug interactions and drug-food interactions in patients with multiple sclerosis. Ther. Adv. Chronic Dis. 2022, 13, 20406223221108391. [Google Scholar] [CrossRef]
- Bachmann, P.; Frahm, N.; Debus, J.L.; Mashhadiakbar, P.; Langhorst, S.E.; Streckenbach, B.; Baldt, J.; Heidler, F.; Hecker, M.; Zettl, U.K. Prevalence and Severity of Potential Drug-Drug Interactions in Patients with Multiple Sclerosis with and without Polypharmacy. Pharmaceutics 2022, 14, 592. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef]
- Amin, M.; Hersh, C.M. Updates and advances in multiple sclerosis neurotherapeutics. Neurodegener. Dis. Manag. 2023, 13, 47–70. [Google Scholar] [CrossRef]
- Lee, A. Ublituximab: First Approval. Drugs 2023, 83, 455–459. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2017, arXiv:1412.6980. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Frahm, N.; Hecker, M.; Langhorst, S.E.; Mashhadiakbar, P.; Haker, M.C.; Zettl, U.K. The risk of polypharmacy, comorbidities and drug-drug interactions in women of childbearing age with multiple sclerosis. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420969501. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.; Pollack, C.; Butkerait, P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther. Clin. Risk Manag. 2015, 11, 1061–1075. [Google Scholar] [CrossRef]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Pharmacogenomics of Drug Metabolizing Enzymes and Transporters: Relevance to Precision Medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef]
- Holstiege, J.; Akmatov, M.K.; Klimke, K.; Dammertz, L.; Kohring, C.; Marx, C.; Frahm, N.; Peters, M.; Ellenberger, D.; Zettl, U.K.; et al. Trends in administrative prevalence of multiple sclerosis and utilization patterns of disease modifying drugs in Germany. Mult. Scler. Relat. Disord. 2022, 59, 103534. [Google Scholar] [CrossRef]
- Wundes, A.; Kraft, G.H.; Bowen, J.D.; Gooley, T.A.; Nash, R.A. Mitoxantrone for worsening multiple sclerosis: Tolerability, toxicity, adherence and efficacy in the clinical setting. Clin. Neurol. Neurosurg. 2010, 112, 876–882. [Google Scholar] [CrossRef]
- Espinola-Klein, C. When and How to Combine Antiplatelet and Anticoagulant Drugs? Hamostaseologie 2022, 42, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Mathews, J. Cladribine Tablets for Relapsing-Remitting Multiple Sclerosis: A Clinician’s Review. Neurol. Ther. 2022, 11, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Han, Q.; Song, H.; Zi, J.; Ma, J.; Ge, Z. Efficacy and toxicity of cladribine for the treatment of refractory acute myeloid leukemia: A meta-analysis. Drug Des. Dev. Ther. 2019, 13, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Criscuolo, M.; Broccoli, A.; Piciocchi, A.; Varettoni, M.; Galli, E.; Anastasia, A.; Cantonetti, M.; Trentin, L.; Kovalchuk, S.; et al. Long-term follow-up of cladribine treatment in hairy cell leukemia: 30-year experience in a multicentric Italian study. Blood Cancer J. 2022, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Siess, W. Bleeding by Bruton Tyrosine Kinase-Inhibitors: Dependency on Drug Type and Disease. Cancers 2021, 13, 1103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Schiller, H.; Huth, F.; Schuhler, C.; Drollmann, A.; Kaul, M.; Woessner, R.; Shah, B.; Weis, W.; End, P. Novel Bruton’s tyrosine kinase inhibitor remibrutinib: Assessment of drug-drug interaction potential as a perpetrator of cytochrome P450 enzymes and drug transporters and the impact of covalent binding on possible drug interactions. Eur. J. Pharm. Sci. 2022, 172, 106155. [Google Scholar] [CrossRef]
- Miller, A.E. An updated review of teriflunomide’s use in multiple sclerosis. Neurodegener. Dis. Manag. 2021, 11, 387–409. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, S.F. Synthetic and natural compounds that interact with human cytochrome P450 1A2 and implications in drug development. Curr. Med. Chem. 2009, 16, 4066–4218. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, G.; Maarbjerg, S.; Truini, A. Trigeminal neuralgia secondary to multiple sclerosis: From the clinical picture to the treatment options. J. Headache Pain 2019, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. Carbamazepine-provoked hepatotoxicity and possible aetiopathological role of glutathione in the events. Retrospective review of old data and call for new investigation. Advers. Drug React. Toxicol. Rev. 2002, 21, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef]
- Domingues, R.B.; Kuster, G.W.; Aquino, C.C. Treatment of trigeminal neuralgia with low doses of topiramate. Arq. Neuropsiquiatr. 2007, 65, 792–794. [Google Scholar] [CrossRef]
- Bendtsen, L.; Zakrzewska, J.M.; Heinskou, T.B.; Hodaie, M.; Leal, P.R.L.; Nurmikko, T.; Obermann, M.; Cruccu, G.; Maarbjerg, S. Advances in diagnosis, classification, pathophysiology, and management of trigeminal neuralgia. Lancet Neurol. 2020, 19, 784–796. [Google Scholar] [CrossRef]
- Raebel, M.A. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc. Ther. 2012, 30, e156–e166. [Google Scholar] [CrossRef]
- Bird, S.T.; Pepe, S.R.; Etminan, M.; Liu, X.; Brophy, J.M.; Delaney, J.A. The association between drospirenone and hyperkalemia: A comparative-safety study. BMC Clin. Pharmacol. 2011, 11, 23. [Google Scholar] [CrossRef]
- Mallareddy, M.; Hanes, V.; White, W.B. Drospirenone, a new progestogen, for postmenopausal women with hypertension. Drugs Aging 2007, 24, 453–466. [Google Scholar] [CrossRef]
- Johnson, B.A.; Seneviratne, C. Alcohol-medical drug interactions. Handb. Clin. Neurol. 2014, 125, 543–559. [Google Scholar] [CrossRef]
- Papasouliotis, O.; Mitchell, D.; Girard, P.; Dangond, F.; Dyroff, M. Determination of a clinically effective evobrutinib dose: Exposure-response analyses of a phase II relapsing multiple sclerosis study. Clin. Transl. Sci. 2022, 15, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Arnold, D.L.; Vermersch, P.; Bar-Or, A.; Fox, R.J.; Matta, A.; Turner, T.; Wallström, E.; Zhang, X.; Mareš, M.; et al. Tolebrutinib Phase 2b Study Group. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: A phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Liu, X.; Ha, J.; Ai, L.; Li, Z. Quantification of orelabrutinib in human plasma and cerebrospinal fluid by liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2023, 1221, 123680. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, Y.; Won, J.H.; Mi Oh, J.; Lee, H. An annotated corpus from biomedical articles to construct a drug-food interaction database. J. Biomed. Inform. 2022, 126, 103985. [Google Scholar] [CrossRef] [PubMed]
- Osuala, E.C.; Ojewole, E.B. Knowledge, attitudes and practices of healthcare professionals regarding drug-food interactions: A scoping review. Int. J. Pharm. Pract. 2021, 29, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Comi, G.; Palace, J.; Siever, A.; Gottschalk, R.; Bijarnia, M.; von Rosenstiel, P.; Tomic, D.; Kappos, L.; FIRST Study Investigators. Assessment of cardiac safety during fingolimod treatment initiation in a real-world relapsing multiple sclerosis population: A phase 3b, open-label study. J. Neurol. 2014, 261, 267–276. [Google Scholar] [CrossRef]
- Türk, D.; Fuhr, L.M.; Marok, F.Z.; Rüdesheim, S.; Kühn, A.; Selzer, D.; Schwab, M.; Lehr, T. Novel models for the prediction of drug-gene interactions. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 1293–1310. [Google Scholar] [CrossRef]
- Bechtold, B.; Clarke, J. Multi-factorial pharmacokinetic interactions: Unraveling complexities in precision drug therapy. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 397–412. [Google Scholar] [CrossRef]
- Lee, G.; Park, C.; Ahn, J. Novel deep learning model for more accurate prediction of drug-drug interaction effects. BMC Bioinform. 2019, 20, 415. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Statistics |
|---|---|
| Sex, n (%) | |
| Women | 441 (70.3) |
| Men | 186 (29.7) |
| Age (years), mean ± SD | 48.6 ± 13.3 |
| Disease duration (years), median (range) | 10 (0–52) |
| Disease course, n (%) | |
| CIS | 27 (4.3) |
| RRMS | 388 (61.9) |
| SPMS | 154 (24.6) |
| PPMS | 58 (9.3) |
| EDSS score, mean ± SD | 3.6 ± 2.1 |
| Comorbidity, n (%) | |
| Yes | 443 (70.7) |
| No | 184 (29.3) |
| Number of medications taken, mean ± SD | |
| Total | 5.3 ± 3.3 |
| Rx | 4.2 ± 3.0 |
| OTC | 1.1 ± 1.3 |
| DMD use, n (%) | |
| Yes | 389 (62.0) |
| No | 238 (38.0) |
| Drug | Type | DrugBank Accession | DDIs in DrugBank 1 |
|---|---|---|---|
| DMDs | |||
| Alemtuzumab | monoclonal antibody | DB00087 | 821 |
| Azathioprine | small molecule | DB00993 | 1276 |
| Cladribine | small molecule | DB00242 | 707 |
| Dimethyl fumarate | small molecule | DB08908 | 340 |
| Diroximel fumarate | small molecule | DB14783 | 342 |
| Fingolimod | small molecule | DB08868 | 560 |
| Glatiramer acetate | peptides | DB05259 | 340 |
| Interferon beta-1a | recombinant protein | DB00060 | 203 |
| Interferon beta-1b | recombinant protein | DB00068 | 643 |
| Mitoxantrone | small molecule | DB01204 | 653 |
| Monomethyl fumarate | small molecule | DB14219 | 340 |
| Natalizumab | monoclonal antibody | DB00108 | 651 |
| Ocrelizumab | monoclonal antibody | DB11988 | 650 |
| Ofatumumab | monoclonal antibody | DB06650 | 685 |
| Ozanimod | small molecule | DB12612 | 589 |
| Peginterferon beta-1a | recombinant protein | DB09122 | 544 |
| Ponesimod | small molecule | DB12016 | 965 |
| Siponimod | small molecule | DB12371 | 1318 |
| Teriflunomide | small molecule | DB08880 | 782 |
| Ublituximab | monoclonal antibody | DB11850 | 360 |
| BTKis | |||
| Evobrutinib | small molecule | DB15170 | 0 |
| Fenebrutinib | small molecule | DB14785 | 0 |
| Orelabrutinib | small molecule | DB16272 | 0 |
| Remibrutinib | small molecule | DB16852 | 0 |
| Tolebrutinib | small molecule | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hecker, M.; Frahm, N.; Zettl, U.K. Update and Application of a Deep Learning Model for the Prediction of Interactions between Drugs Used by Patients with Multiple Sclerosis. Pharmaceutics 2024, 16, 3. https://doi.org/10.3390/pharmaceutics16010003
Hecker M, Frahm N, Zettl UK. Update and Application of a Deep Learning Model for the Prediction of Interactions between Drugs Used by Patients with Multiple Sclerosis. Pharmaceutics. 2024; 16(1):3. https://doi.org/10.3390/pharmaceutics16010003
Chicago/Turabian StyleHecker, Michael, Niklas Frahm, and Uwe Klaus Zettl. 2024. "Update and Application of a Deep Learning Model for the Prediction of Interactions between Drugs Used by Patients with Multiple Sclerosis" Pharmaceutics 16, no. 1: 3. https://doi.org/10.3390/pharmaceutics16010003